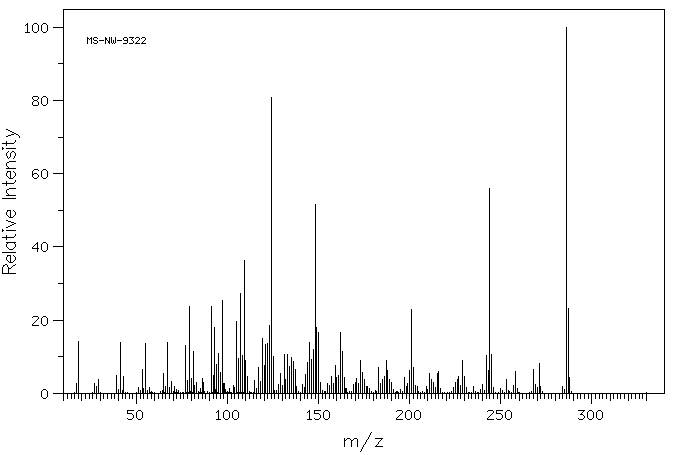
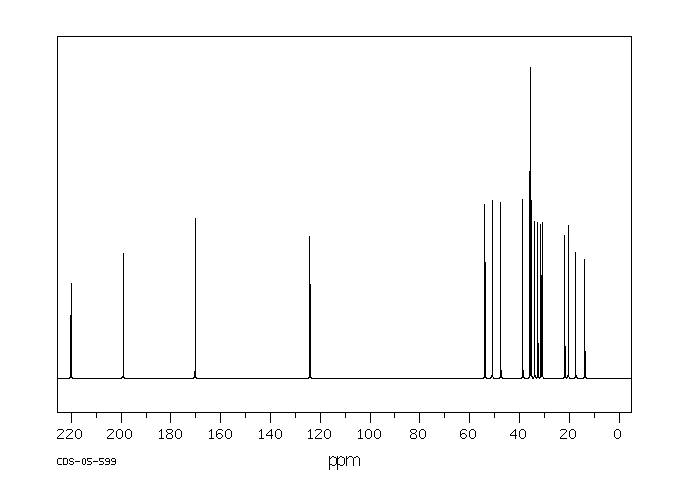
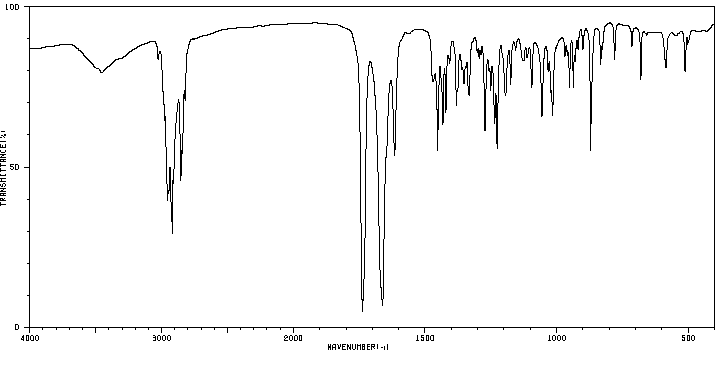
A comparative study was performed to assess the metabolism of the androgen precursor androstenedione (AD) in two gastropod species from the Muricidae family: Bolinus brandaris and Hexaplex trunculus. AD was mainly converted to 5alpha-dihydrotestosterone by microsomal fractions isolated from Bolinus brandaris, whereas it was primarily metabolized to testosterone by Hexaplex trunculus. Sex differences in the metabolism of AD were only detected in Bolinus brandaris and attributed to higher 5alpha-reductase activity in males. Thereafter, the effect of the organotin compounds, tributyltin (TBT) and triphenyltin (TPT), on the metabolism of AD was investigated. A significant interference was only detected in females, and differences between the modes of action of the two compounds were observed: TPT was a strong inhibitor of 5alpha-reductase activity in B. brandaris at a concentration as low as 100 nM whereas only TBT (10 uM) altered the metabolism of AD in H. trunculus by increasing the activity 17beta-hydroxysteroid dehydrogenase (17beta-HSD). Thus, this work shows that the metabolism of the androgen precursor AD strongly differs among gastropod species, both in terms of activity and metabolic profile, and further demonstrates the ability of TBT and TPT to interfere with key enzymatic pathways involved in androgen synthesis.
Bone is a target organ of androgens. The mechanism by which these steroids exert their action within bone cells is still poorly understood. The metabolism of androstenedione, the major circulating androgen in women, was, therefore, assessed in osteoblast-like bone cells cultured from bone of 16 postmenopausal women (mean age, 69 yr; range, 56-80) and 3 elderly men (mean age, 71 yr; range, 69-73) undergoing total hip replacement. Each cell strain was incubated under standardized conditions with varying concentrations of [1,2,6,7- (3)H]androstenedione (0.05-5 uM). In every instance 5 alpha-reduced metabolites and 17 beta-hydroxysteroids were formed. There was no correlation between the volumetric density of the resected bone and androstenedione metabolism of the corresponding cultured bone cell strains. The apparent Km for the 5 alpha-reductase activity (sum of androstanedione and dihydrotestosterone) of all 19 cell strains was 0.7 +/- 0.1 uM (mean +/- SEM), and the apparent Km for 17 beta- hydroxysteroid dehydrogenase (sum of testosterone and dihydrotestosterone) was 2.3 +/- 0.8 uM (mean +/- SEM), values similar to those reported for other androgen target organs. Our results demonstrate that human osteoblast-like cells have the capacity to transform androstenedione into the more potent biological androgens testosterone and dihydrotestosterone. Since the Km values of both 5 alpha-reductase and 17 beta-hydroxysteroid dehydrogenase exceed the serum androstenedione concentration, the formation of testosterone and dihydrotestosterone appears to be mainly a function of substrate availability.
Up-regulation of aromatase expression in endometrial cells disseminated into the peritoneal cavity may enhance their survival via local estrogen synthesis, which may lead to endometriosis. The factors that mediate induction of aromatase in the endometrium are not well defined, but increased expression of steroidogenic factor (SF)-1 may play a role. The objective of the study was to determine whether androstenedione (A4), the predominant sex steroid in peritoneal fluid, regulates endometrial aromatase expression. This was a cell/tissue culture study ... conducted at an academic center. Quantitative real-time PCR, HPLC, and chromatin immunoprecipitation were used in this study. Treatment of cultured human endometrial explants and stromal cells with A4 (10 nm) significantly up-regulated expression of aromatase mRNA transcripts containing exon IIa at their 5'-ends. In endometrial stromal cells and the human endometrial surface epithelial (HES) cell line, induction of aromatase mRNA by A4 was associated with increased expression of SF-1. In HES cells, tritiated A4 was metabolized to estradiol, testosterone (T), dihydrotestosterone, and androstanediol. Both estradiol and T, but not nonaromatizable androgens, up-regulated aromatase and SF-1 mRNA in HES cells. Chromatin immunoprecipitation revealed that A4 enhanced recruitment of SF-1 to its response element (-136 bp) upstream of CYP19 exon IIa. This, together with the findings that both estrogen receptor antagonist, ICI 182,780, and aromatase inhibitor, fadrozole, suppressed A4 and T induction of aromatase and SF-1 mRNA, indicates that the inductive effects of A4 and T are mediated by their conversion to estrogens. Exposure of endometrial cells to A4 may enhance CYP19 gene expression through its aromatization to estrogens.
Androstenedione is synthesized in the adrenal gland and gonads from dehydroepiandrosterone. It is metabolized by the enzyme 17 beta-hydroxy steroid dehydrogenase to testosterone, and by the aromatase enzyme complex to estrone. Estrone is metabolized to estradiol.
Androstenedione is distributed to various tissues of the body and is metabolized to testosterone and estrone. The amount of testosterone produced per given dose of androstenedione appears to vary. Typically, a greater increase in serum testosterone is found in women compared to men, following intake of oral androstenedione.
IDENTIFICATION AND USE: Androstenedione is a synthetic analog of androgens, formerly used as a dietary supplement. HUMAN STUDIES: There are cases of androstenedione-induced impotence and severe oligospermia. The effects of androstenedione supplementation on the hormonal profile of 10 males and its interaction with resistance exercise have been studied. Exercise elevated testosterone, with no difference between conditions. Exercise in the supplemented condition significantly elevated plasma estradiol by approximately 83% for 90 min. Androstenedione supplementation, thus, is unlikely to provide male athletes with any anabolic benefit and, with heavy resistance exercise, elevates estrogen. Other study suggested that oral androstenedione, when given in dosages of 300 mg/day, increased serum testosterone and estradiol concentrations in some healthy men. However, supplementation does result in significant increases in estrogen-related compounds, dehydroepiandrosterone sulfate concentrations, down-regulation in testosterone synthesis, and unfavorable alterations in blood lipid and coronary heart disease risk profiles of men aged 35 to 65 years. ANIMAL STUDIES: Androstenedione did not reveal a skin sensitization potential when tested in a guinea pig maximization test with a concentration of 5% for intradermal induction and 25% for epicutaneous induction and challenge following. Eye irritation was reported in rabbits. A single oral dose of androstenedione to male rats caused mortality at 1000 mg/kg, whereas animals survived at 500 mg/kg. Compound-related clinical signs were apathy, disturbance of gait and squatting position. At the lethal dose, additional signs included prone position, unconsciousness, disturbance in respiration and increased diuresis. The once daily oral administration of androstenedione to male and female rats over 4 weeks at doses of 0, 15, 50 and 150 mg/kg led to dose-dependent effects in females, such as increased body weight, atrophy of uterus, cervix, pituitary gland and adrenals, as well as increased numbers of erythrocytes and increased hemoglobin. For males alterations in thymus were reported. These effects were regarded to represent endocrine effects typical for a steroid hormone. Infusion of androstenedione to pregnant monkeys resulted in the premature occurrence of labor-type myometrial activity and increases in maternal plasma estrogen, oxytocin and amnion fibronectin concentrations similar to those measured at normal-term labor. In rats, androstenedione had no specific effect on the development of individual bones or soft tissues. Androstenedione did not show a mutagenic potential in a bacterial reverse mutation assay according to OECD TG 471 (Ames test in S. typhimurium TA98, TA1537, TA100, TA102, TA1535) when tested up to the highest recommended dose level of 5.0 mg/plate in the absence or presence of metabolic activation. ECOTOXICITY STUDIES: Exposure to androstenedione via water can cause masculinization of adult female mosquitofish in a relatively short period of time. Exogenous androstenedione, at environmentally relevant concentrations, can significantly modulate the reproductive physiology of the fathead minnows in a sex-specific manner.
Androstenedione is converted to testosterone and estrogen, and when taken in sufficient quantities androstenedione can cause unwanted masculinizing and feminizing effects. Androstenedione is considered an androgenic steroid precursor because testosterone is an androgen or male hormone. In males, conversion of androstenedione to testosterone requires the enzyme 17β-hydroxysteroid dehydrogenase. In females, conversion of androstenedione to estrogen (e.g., estrone and estradiol) requires the enzyme aromatase.
来源:Toxin and Toxin Target Database (T3DB)
毒理性
致癌物分类
对人类不具有致癌性(未被国际癌症研究机构IARC列名)。
No indication of carcinogenicity to humans (not listed by IARC).
Androstenedione was legally defined as an anabolic steroid by the FDA in 2004. It poses significant health risks commonly associated with steroids. The side effects for men include breast development, behavioral changes, heart disease, and more. Side effects for women are similar to the side effects from anabolic steroids in that their voices will deepen and they may grow facial hair since both occur from an increase level of testosterone. Another side effect of androstenedione is male pattern baldness. The main psychological side effect of androstenedione is depression. Mood swings are also common. Chronically high levels of androstenedione are associated with male pseudohermaphrodism with gynecomastia, Adrenal Hyperplasia Type 3 and Aromatase deficiency.
Absorption of /orally administered androstenedione/ appears variable, but some absorption does occur. Androstenedione is distributed to various tissues of the body ... .
/MILK/ It is not known whether anabolic steroids are distributed into breast milk. Problems in humans have not been documented. Women who take anabolic steroids should not breast feed. /Anabolic Steroids/
The present invention relates to compounds or pharmaceutically-acceptable salts thereof, processes for preparing them, pharmaceutical compositions containing them and their use in therapy. The invention particularly relates to compounds that are polo-like kinase (PLKs) inhibitors useful for the treatment of disease states mediated by PLK, especially PLK4, in particular such compounds that are useful in the treatment of pathological processes which involve an aberrant cellular proliferation, such as tumour growth, rheumatoid arthritis, restenosis and atherosclerosis.
The present invention relates to compounds of formula I
and pharmaceutically acceptable salts. These compounds can act as potential MEK inhibitors in the treatment of hyperproliferative diseases, like cancer and inflammation. The present invention also reveals methods of preparation thereof.
Synthesis, stereochemistry and cytotoxic activity of novel steroidal 16-spiro-1,3,2-dioxaphosphorinanes
作者:János Wölfling、Piroska Kovács-Pénzes、István Zupkó、Gyula Schneider、Éva Frank
DOI:10.1016/j.molstruc.2012.01.013
日期:2012.4
Abstract The epimeric pairs a and b of novel steroidal 16-spiro-dioxaphosphorinanes 4–8 were synthetized via the phosphorylation of 16,16-bis(hydroxymethyl)androst-4-ene-3,17-dione (2) and their stereostructures were investigated by NMR methods. The dioxaphosphorinane moiety exists mainly as one of the possible chair conformers or as a chair–twist equilibrium in solution as a consequence of the rigidity
摘要 通过 16,16-双(羟甲基)androst-4-ene-3,17-dione (2) 的磷酸化合成了新型甾体 16-spiro-dioxaphosphorinanes 4-8 的差向异构对 a 和 b,其立体结构为用核磁共振方法研究。由于甾烷骨架的刚性,二恶磷烷部分主要作为可能的椅子构象异构体之一或作为溶液中的椅子扭曲平衡存在。构象异构体的贡献很大程度上取决于 P 原子的构型和取代基的立体电子特性。结构相关产物的抗增殖活性在体外用 MTT 测定法对三种恶性人类细胞系(HeLa、MCF7 和 A431)进行了测定。
[EN] 3-PHOSPHOGLYCERATE DEHYDROGENASE INHIBITORS AND USES THEREOF<br/>[FR] INHIBITEURS DE LA 3-PHOSPHOGLYCÉRATE DÉSHYDROGÉNASE ET LEURS UTILISATIONS
申请人:RAZE THERAPEUTICS INC
公开号:WO2017156181A1
公开(公告)日:2017-09-14
The present invention provides compounds, compositions thereof, and methods of using the same.