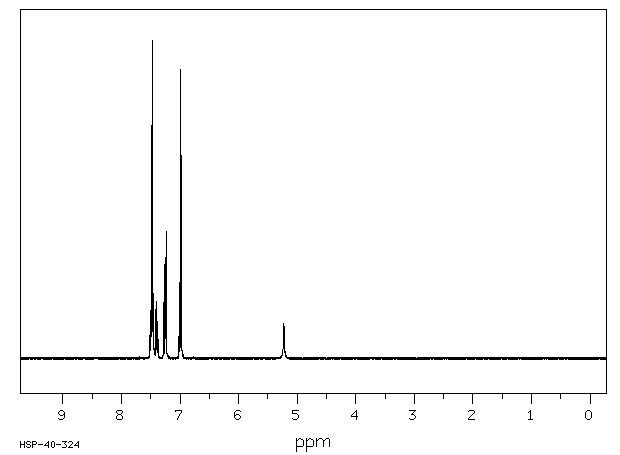
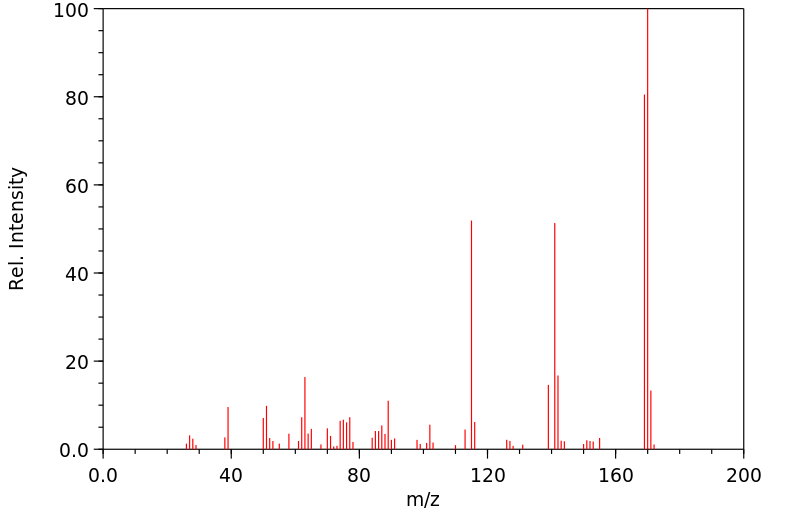
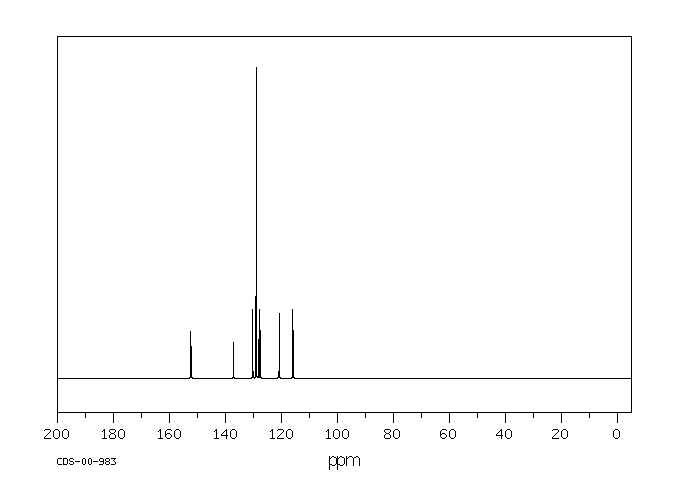
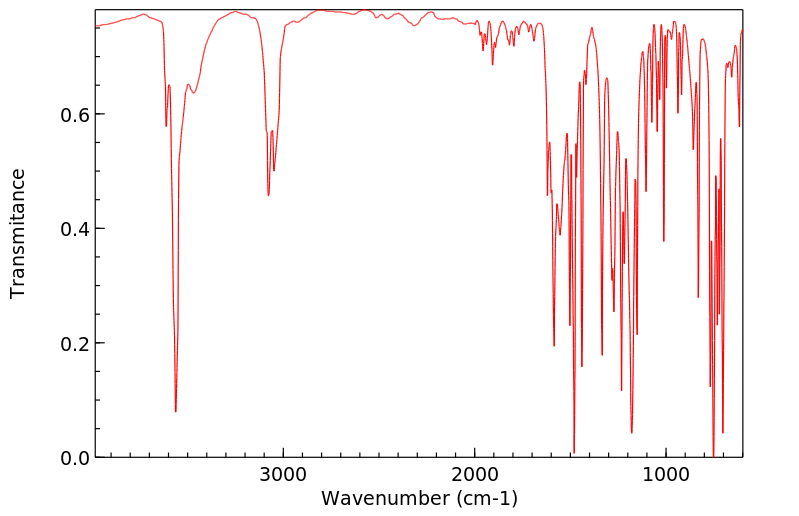
Ortho-phenylphenol (OPP) was well absorbed in the male B6C3F1 mouse, with 84 and 98% of the administered radioactivity recovered in the 0-48-hr urine of animals administered a single oral dose of 15 or 800 mg/kg respectively. High absorption and rapid elimination were also seen in the female and male F344 rat with 86 and 89% respectively of a single oral dose (27-28 mg/kg) found in the urine in 24 hr. OPP was also rapidly eliminated from human volunteers following dermal exposure for 8 hr (0.006 mg/kg), with 99% of the absorbed dose in the urine in 48 hr.. Sulfation of OPP was found to be the major metabolic pathway at low doses in all three species, accounting for 57, 82 and 69% of the urinary radioactivity in the male mouse (15 mg/kg, po), male rat (28 mg/kg, po) and male human volunteers (0.006 mg/kg, dermal). OPP-glucuronide was also present in all species, representing 29, 7 and 4% of the total urinary metabolites in the low dose groups of mouse, rat and human volunteers respectively. Conjugates of 2-phenylhydroquinone (PHQ) in these single-dose studies accounted for 12, 5 and 15% of the dose in mouse, rat and human, respectively. Little or no free OPP was found in any species. No free PHQ or PBQ was found in the mouse, rat or human (LOD = 0.1-0.6%). A novel metabolite, the sulfate conjugate of 2,4'-dihydroxybiphenyl, was identified in rat and man, comprising 3 and 13% of the low dose respectively. Dose-dependent shifts in metabolism were seen in the mouse for conjugation of parent OPP, indicating saturation of the sulfation pathway. Dose-dependent increases in total PHQ were also observed in mouse. This study was initiated to elucidate a mechanistic basis for the difference in carcinogenic potential for OPP between rat and mouse. However, the minor differences seen in the metabolism of OPP in these two species do not appear to account for the differences in urinary bladder toxicity and tumor response between mouse and rat.
(14)C-ortho-Phenylphenol was applied onto the skin of the forearm of 6 volunteers for 8 hr at a dose of 0.4 mg/person (0.006 mg/kg bw). ... Sulfation was the major metabolic pathway, accounting for 69% of the metabolites, while conjugates of 2-phenylhydroquinone accounted for 15%. Little or no free ortho-phenylphenol was present in the urine, & no free 2-phenylhydroquinone or 2-phenyl-1,4-benzoquinone was detected.
ortho-Phenylphenol was converted to phenylhydroquinone by microsomal cytochrome P450 in vitro. Phenylhydroquinone was oxidized to phenylquinone by cumene hydroperoxide-supported microsomal cytochrome P450, & phenylquinone was reduced back to phenylhydroquinone by cytochrome P450 reductase, providing direct evidence of redox cycling of ortho-phenylphenol.
Evaluation: There is inadequate evidence in humans for the carcinogenicity of ortho-phenylphenol and sodium ortho-phenylphenate. Overall evaluation: ortho-Phenylphenol is not classifiable as to its carcinogenicity to humans (Group 3). Sodium ortho-phenylphenate is possibly carcinogenic to humans (Group 2B).
The pharmacokinetics and metabolism of uniformly labeled 14C/13C-ortho-phenylphenol (OPP) were followed in six human male volunteers given a single 8 hr dermal dose of 6 ug OPP/kg body weight formulated as a 0.4% (w/v) solution in isopropyl alcohol. The application site was covered with a non-occlusive dome allowing free movement of air, but preventing the loss of radioactivity due to physical contact. At 8 hr post-exposure the non-occlusive dome was removed, the dose site was wiped with isopropyl alcohol containing swabs and the skin surface repeatedly stripped with tape. Blood specimens, urine, and feces were collected from each volunteer over a 5 day post-exposure period and were analyzed for radioactivity and metabolites (urine only). Following dermal application, peak plasma levels of radioactivity were obtained within 4 hr post-exposure and rapidly declined with virtually all of the absorbed dose rapidly excreted into the urine within 24 hr post-exposure. A one-compartment pharmacokinetic model was used to describe the time-course of OPP absorption and clearance in male human volunteers. Approximately 43% of the dermally applied dose was absorbed through the skin with an average absorption half-life of 10 hr. Once absorbed the renal clearance of OPP was rapid with an average half-life of 0.8 hr. The rate limiting step for renal clearance was the relatively slower rate of dermal absorption; therefore the pharmacokinetics of OPP in humans was described by a 'flip-flop' single compartment model. Overall, the pharmacokinetics were similar between individuals, and the model parameters were in excellent agreement with the experimental data. Approximately 73% of the total urinary radioactivity was accounted for as free OPP, OPP-sulfate and OPP-glucuronide conjugates. The sulfate conjugate was the major metabolite (approximately 69%). Therefore, total urinary OPP equivalents (acid-labile conjugates+free OPP) can be used to estimate the systemically absorbed dose of OPP. The rapid excretion of OPP and metabolites into the urine following dermal exposure indicates that OPP is unlikely to accumulate in humans upon repeated exposure ...
Male F344 rats were treated with 0, 15, 50, 125, 250, 500, 1000 mg/kg of ortho-phenylphenol (OPP) and its radiocarbon analogue via oral gavage. The dosed rats were euthanized after 24 hr, and the proteins were extracted from the liver, kidney, and bladder. The amount of radioactivity associated with the extracted protein was quantified ... Protein binding in liver and kidney exhibited a linear or modest curvilinear relationship over the dose range studied. In the urinary bladder, however, a pronounced nonlinear relationship between protein adduct levels and administered dose was observed. The measured protein adduct levels were in agreement with the predicted concentrations of phenylbenzoquinone based on a proposed mechanism involving free phenylhydroquinone autoxidation in the urine. Unlike protein binding, DNA adducts measured from the same bladder samples did not show a significant difference from the control group ...
The validity of in vitro and in vivo methods for the prediction of percutaneous penetration in humans was assessed using the fungicide ortho-phenylphenol (OPP) (log Po/w 3.28, MW 170.8, solubility in water 0.7 g/L). In vivo studies were performed in rats and human volunteers, applying the test compound to the dorsal skin and the volar aspect of the forearm, respectively. In vitro studies were performed using static diffusion cells with viable full-thickness skin membranes (rat and human), nonviable epidermal membranes (rat and human), and a perfused pig ear model. For the purpose of conducting in vitro/in vivo comparisons, standardized experimental conditions were used with respect to dose (120 ug OPP/sq cm), vehicle (60% aqueous ethanol), and exposure duration (4 hr). In human volunteers, the potentially absorbed dose (amount applied minus dislodged) was 105 ug/sq cm, while approximately 27% of the applied dose was excreted with urine within 48 hr. In rats these values were 67 ug/sq cm and 40%, respectively. In vitro methods accurately predicted human in vivo percutaneous absorption of OPP on the basis of the potential absorbed dose. With respect to the other parameters studied (amount systemically available, maximal flux), considerable differences were observed between the various in vitro models ...
1.周国泰,化学危险品安全技术全书,化学工业出版社,1997 2.国家环保局有毒化学品管理办公室、北京化工研究院合编,化学品毒性法规环境数据手册,中国环境科学出版社.1992 3.Canadian Centre for Occupational Health and Safety,CHEMINFO Database.1998 4.Canadian Centre for Occupational Health and Safety, RTECS Database, 1989
亲核芳香取代 (S N Ar) 是一种通过用亲核试剂取代离去基团将杂原子结合到芳香环中的强大策略,但这种方法仅限于缺电子芳烃。我们现在已经建立了一种通过催化 S N Ar 反应获取苯酚和苯基烷基醚的可靠方法。该方法适用于广泛的富电子和中性芳基氟化物,它们在经典的 S N Ar 条件下呈惰性。虽然 S N的机制假设涉及金属芳烃配合物的 Ar 反应涉及逐步途径(添加后消除),支持该假设的实验数据仍在探索中。机械研究和 DFT 计算表明了一个逐步或逐步的能量分布。值得注意的是,我们分离了铑 η 5 -环己二烯基复合中间体,其具有带有亲核试剂和离去基团的 sp 3 -杂化碳。
A General and Mild Copper-Catalyzed Arylation of Diethyl Malonate
摘要:
GRAPHICSA general method for the synthesis of alpha-aryl malonates is described. The coupling of an aryl iodide and diethyl malonate in the presence of Cs2CO3 and catalytic amounts of copper(I) iodide and 2-phenylphenol affords the alpha-aryl malonate in good to excellent yields. The mild reaction conditions and high levels of functional group compatibility make this an attractive synthetic alternative to previous methods.
Heterocyclic derivatives for the treatment of cancer and other proliferative diseases
申请人:——
公开号:US20020143182A1
公开(公告)日:2002-10-03
The invention relates to certain heterocyclic compounds useful for the treatment of cancer and other diseases, having the Formula (I):
1
wherein:
(a) m is an integer 0 or 1;
(b) R
12
is an alkyl, a substituted alkyl, a cycloalkyl, a substituted cycloalkyl, a heterocyclic, a substituted heterocyclic, a heteroaryl, a substituted heteroaryl, an aryl or a substituted aryl residue;
(c) Ar
3
is an aryl, a substituted aryl, a heteroaryl or a substituted heteroaryl residue;
(d) Ar
4
is an aryl, a substituted aryl, a heteroaryl or a substituted heteroaryl residue;
(e) R
5
is hydrogen, hydroxy, alkyl or substituted alkyl;
(f) - - - - - represents a bond present or absent; and
(g) W, X, Y and Z are independently or together C(O)—, C(S), S, O, or NH; or a pharmaceutically acceptable salt thereof.
Chiral 2-(2-hydroxyaryl)alcohols (HAROLs) with a 1,4-diol scaffold as a new family of ligands and organocatalysts
作者:Ömer Dilek、Mustafa A. Tezeren、Tahir Tilki、Erkan Ertürk
DOI:10.1016/j.tet.2017.11.054
日期:2018.1
Efficient and modular syntheses of chiral 2-(2-hydroxyaryl)alcohols (HAROLs), novel 1,4-diols carrying one phenolic and one alcohol hydroxyl group, have been developed which led to generation of a small library of structurally diverse HAROLs in enantiomerically pure form. Of the different HAROLs examined, a HAROL based on the indan backbone exhibited the highest activity and enantioselectivity in the
高效和模块化的手性2-(2-羟基芳基)醇(HAROLs)的合成,带有一个酚和一个醇羟基的新型1,4-二醇已被开发出来,这导致在对映异构体中生成结构多样的HAROLS小文库纯形式。在考察的不同HAROL中,基于茚满骨架的HAROL在Ti(O i Pr)4(y高达97%, 88%ee)并在三田膦的促进下在Morita-Baylis-Hillman反应中作为氢键供体有机催化剂发挥作用。
Novel thienylpyridylcarboxamides of the formula (I)
The present application is also directed to a plurality of processes for preparing these compounds and their use for controlling unwanted microorganisms, and also novel intermediates and their preparation.