
6-氯鸟嘌呤核苷 | 2004-07-1
中文名称
6-氯鸟嘌呤核苷
中文别名
2-氨基-6-氯-9-(β-D-呋喃核糖基)嘌呤;6-氯腺苷;2-氨基-6-氯嘌呤核苷;6-氯鸟嘌呤核甙;6-氯鸟苷;6-氯鸟嘌呤苷
英文名称
2-Amino-6-chloropurine riboside
英文别名
2-amino-6-chloro-9-(β-D-ribofuranosyl)purine;6-Chloroguanosine;(2R,3R,4S,5R)-2-(2-amino-6-chloropurin-9-yl)-5-(hydroxymethyl)oxolane-3,4-diol

CAS
2004-07-1
化学式
C10H12ClN5O4
mdl
——
分子量
301.689
InChiKey
TXWHPSZYRUHEGT-UUOKFMHZSA-N
BEILSTEIN
——
EINECS
——
-
物化性质
-
计算性质
-
ADMET
-
安全信息
-
SDS
-
制备方法与用途
-
上下游信息
-
文献信息
-
表征谱图
-
同类化合物
-
相关功能分类
-
相关结构分类
物化性质
-
熔点:165-167 °C (dec.)(lit.)
-
沸点:729.9±70.0 °C(Predicted)
-
密度:1.8359 (rough estimate)
-
溶解度:可溶于二甲基亚砜、甲醇
计算性质
-
辛醇/水分配系数(LogP):-0.1
-
重原子数:20
-
可旋转键数:2
-
环数:3.0
-
sp3杂化的碳原子比例:0.5
-
拓扑面积:140
-
氢给体数:4
-
氢受体数:8
安全信息
-
危险品标志:Xi
-
安全说明:S24/25
-
危险类别码:R36/37/38
-
WGK Germany:3
-
海关编码:29349990
-
危险品运输编号:OTH
-
危险性防范说明:P261,P280,P305+P351+P338
-
危险性描述:H302,H315,H319,H332,H335
SDS
上下游信息
-
上游原料
中文名称 英文名称 CAS号 化学式 分子量 —— 2-amino-6-chloro-9-<3,5-bis-O-(tert-butyldimethylsilyl)-β-D-ribofuranosyl>purine 133519-47-8 C22H40ClN5O4Si2 530.215 2,6-二氯嘌呤核苷 2,6-dichloropurine riboside 13276-52-3 C10H10Cl2N4O4 321.12 —— 6-chloro-2-fluoropurine riboside 19083-18-2 C10H10ClFN4O4 304.665 鸟苷 GUANOSINE 118-00-3 C10H13N5O5 283.244 —— 2',3',5'-Tri-O-benzoyl-6-chloroguanosin 3387-63-1 C31H24ClN5O7 614.014 2',3',5'-三乙酰鸟苷 2',3',5'-tri-O-acetyl-guanosine 6979-94-8 C16H19N5O8 409.356 尿嘧啶核苷 uridine 58-96-8 C9H12N2O6 244.204 -
下游产品
中文名称 英文名称 CAS号 化学式 分子量 6-氯-9-(2-O-甲基-beta-D-呋喃核糖基)-9H-嘌呤-2-胺 2-amino-6-chloro-9-[2'-O-methyl-β-D-ribofuranosyl]purine 194034-59-8 C11H14ClN5O4 315.716 —— 2-amino-9-(5-O-tert-butyldimethylsilyl-β-D-ribofuranosyl)-6-chloropurine 413570-50-0 C16H26ClN5O4Si 415.952 —— 2-amino-6-chloro-9-(2-deoxy-β-D-threo-pentofuranosyl)purine 933773-94-5 C10H12ClN5O3 285.69 2-氨基嘌呤核苷 2-amino-9-β-D-ribofuranosylpurine 4546-54-7 C10H13N5O4 267.244 —— 2-amino-6-chloro-9-<3,5-bis-O-(tert-butyldimethylsilyl)-β-D-ribofuranosyl>purine 133519-47-8 C22H40ClN5O4Si2 530.215 —— 2-(2'-amino-6'-chloropurin-9'-yl)-5-hydroxymethyl-3,4-O-isopropylidene-tetrahydrofuran 63629-80-1 C13H16ClN5O4 341.754 —— 2-amino-6-chloro-9-<2,5-bis-O-(tert-butyldimethylsilyl)-β-D-ribofuranosyl>purine 133519-46-7 C22H40ClN5O4Si2 530.215 —— 9-[(2R,3R,4R,5R)-3,4-bis(trimethylsilyloxy)-5-(trimethylsilyloxymethyl)oxolan-2-yl]-6-chloropurin-2-amine 190773-85-4 C19H36ClN5O4Si3 518.235 [(2S,5R)-5-(2-氨基-6-氯嘌呤-9-基)四氢呋喃-2-基]甲醇 2-amino-6-chloro-9-(2,3-dideoxy-β-D-glycero-pentofuranosyl)-9H-purine 122970-35-8 C10H12ClN5O2 269.691 —— 2-amino-6-chloro-9<3-deoxy-2,5-bis-O-(tert-butyldimethylsilyl)-β-D-ribofuranosyl>purine 132121-72-3 C22H40ClN5O3Si2 514.215 2-氨基腺嘌呤核苷 2-aminoadenosine 2096-10-8 C10H14N6O4 282.259 —— 2-amino-6-methylpurine 9-β-D-ribonucleoside 80681-62-5 C11H15N5O4 281.271 —— 2-amino-6-fluoropurine riboside 34798-92-0 C10H12FN5O4 285.235 —— 2-amino-6-chloro-9-(2,3-dideoxy-3-fluoro-β-D-erythro-pentofuranosyl)purine 134379-74-1 C10H11ClFN5O2 287.681 —— 2-amino-9-(5-O-tert-butyldimethylsilyl-2,3-O-thiocarbonyl-β-D-ribofuranosyl)-6-chloropurine 413570-52-2 C17H24ClN5O4SSi 458.013 —— (18)O(6)-Guanosine 3001-34-1 C10H13N5O5 285.244 —— 2-(2'-amino-6'-chloropurin-9'-yl)-5-carboxy-3,4-O-isopropylidene-tetrahydrofuran 936252-99-2 C13H14ClN5O5 355.738 —— 2-amino-6-chloro-9-[3,5-O-(tetraisopropyldisiloxane-1,3-diyl)-β-D-ribofuranosyl]purine 87791-91-1 C22H38ClN5O5Si2 544.198 —— (2R,3R,4S,5R)-2-[6-amino-2-(2-aminoethylamino)purin-9-yl]-5-(hydroxymethyl)oxolane-3,4-diol 77112-18-6 C12H19N7O4 325.327 —— (Z)-2-amino-9-((2R,3R,4S,5R)-3,4-dihydroxy-5-(hydroxymethyl)tetrahydrofuran-2-yl)-1,9-dihydro-6H-purin-6-one oxime 7269-58-1 C10H14N6O5 298.26 —— 1-(2-amino-6-hydroxyamino-purin-9-yl)-β-D-1-deoxy-ribofuranose 7269-58-1 C10H14N6O5 298.258 —— N2-Propyl-2-aminoadenosin 35109-89-8 C13H20N6O4 324.34 —— (2R,3R,4S,5R)-2-(2-amino-6-(methylthio)-9H-purin-9-yl)-5(hydroxymethyl)tetrahydrofuran-3,4-diol —— C11H15N5O4S 313.337 —— (2R,3R,4S,5R)-2-[2-amino-6-(methylamino)-9H-purin-9-yl]-5-(hydroxymethyl)tetrahydrofuran-3,4-diol 5059-59-6 C11H16N6O4 296.286 —— 2-(n-butylamino)adenosine 255716-02-0 C14H22N6O4 338.366 2'-甲氧基鸟苷 2'-O-methylguanosine 2140-71-8 C11H15N5O5 297.271 —— 2-amino-N6-aminoadenosine 6220-31-1 C10H15N7O4 297.274 —— 2-[(4''-hydroxybutyl)amino]adenosine —— C14H22N6O5 354.366 —— 2-(n-amylamino)adenosine —— C15H24N6O4 352.393 —— 2-[(2''-hydroxypropyl)amino]adenosine —— C13H20N6O5 340.339 —— 2-amino-9-(2-deoxy-β-D-ribofuranosyl)purine 3616-24-8 C10H13N5O3 251.245 —— N2-Cyclohexyl-2,6-diaminonebularin 53296-12-1 C16H24N6O4 364.404 —— N6-Methoxy-2-aminoadenosin 55652-73-8 C11H16N6O5 312.285 —— 2-[(2'',3''-dihydroxypropyl)amino]adenosine —— C13H20N6O6 356.338 —— 9-[(2R,3R,4R,5R)-3,4-bis(trimethylsilyloxy)-5-(trimethylsilyloxymethyl)oxolan-2-yl]purin-2-amine 1053614-74-6 C19H37N5O4Si3 483.79 —— (2R,3R,4S,5R)-2-[6-amino-2-(cyclopentylamino)purin-9-yl]-5-(hydroxymethyl)oxolane-3,4-diol 57972-89-1 C15H22N6O4 350.377 N2,N2-二甲基鸟苷 N2,N2-dimethylguanosine 2140-67-2 C12H17N5O5 311.297 6-O-甲基-鸟苷 6-O-methylguanosine 7803-88-5 C11H15N5O5 297.271 —— 2-[(cyclohexylmethyl)amino]adenosine —— C17H26N6O4 378.431 —— CGS 21223 56720-67-3 C18H22N6O4 386.41 —— 2-amino-N6-(3''-aminopropyl)adenosine 313476-82-3 C13H21N7O4 339.354 —— 2-[(m-methylbenzyl)amino]adenosine —— C18H22N6O4 386.41 —— 2-amino-N6-(4''-hydroxybutyl)adenosine —— C14H22N6O5 354.366 —— 2-amino-N6-allyladenosine 26783-43-7 C13H18N6O4 322.324 —— (2R,3R,4S,5R)-2-(6-Amino-2-((3-phenylpropyl)amino)-9H-purin-9-yl)-5-(hydroxymethyl)tetrahydrofuran-3,4-diol 124499-05-4 C19H24N6O4 400.437 —— 2-amino-N6-(2''-hydroxypropyl)adenosine —— C13H20N6O5 340.339 —— 2-[[2''-(p-hydroxyphenyl)ethyl]amino]adenosine 124499-08-7 C18H22N6O5 402.41 —— 2-[2-(4-aminophenyl)ethylamino]adenosine 161536-30-7 C18H23N7O4 401.425 6-O-乙基鸟苷 O-6-Ethylguanosin 39708-01-5 C12H17N5O5 311.297 —— 2,6-diaminopurine riboside-5'-O-[(phosphonomethyl)phosphonic acid] —— C11H18N6O9P2 440.246 2',3',5'-三-O-乙酰-6-氯-2-碘嘌呤核苷 6-chloro-2-iodo-9-(2',3',5'-tri-O-acetyl-β-D-ribofuranosyl)-9H-purine 5987-76-8 C16H16ClIN4O7 538.683 2'-O-甲基腺苷 2'-O-methyl adenosine 2140-79-6 C11H15N5O4 281.271 —— 2-amino-N6-(3''-amino-2''-hydroxypropyl)adenosine —— C13H21N7O5 355.354 2-碘腺苷 2-iodoadenosine 35109-88-7 C10H12IN5O4 393.141 —— 2-{6-Amino-2-[2-(4-methoxy-phenyl)-ethylamino]-purin-9-yl}-5-hydroxymethyl-tetrahydro-furan-3,4-diol 124499-07-6 C19H24N6O5 416.437 —— (2R,3R,4S,5R)-2-[2-amino-6-(cyclopentylamino)-9H-purin-9-yl]-5-(hydroxymethyl)tetrahydrofuran-3,4-diol 120524-28-9 C15H22N6O4 350.377 —— 2-(2-phenylpropylamino)-adenosine 124498-68-6 C19H24N6O4 400.437 —— (2R,3R,4S,5R)-2-[6-amino-2-[(2-hydroxy-2-phenylethyl)amino]purin-9-yl]-5-(hydroxymethyl)oxolane-3,4-diol 124498-53-9 C18H22N6O5 402.41 —— 2-amino-N6-(cyclohexylmethyl)adenosine —— C17H26N6O4 378.431 —— 2-amino-6-(benzylamino)-9-(β-D-ribofuranosyl)purine 26783-32-4 C17H20N6O4 372.384 —— (2R,3R,4S,5R)-2-[2-amino-6-(azetidin-1-yl)purin-9-yl]-5-(hydroxymethyl)oxolane-3,4-diol 1403250-65-6 C13H18N6O4 322.324 —— N6-(β-phenethyl)-2-aminoadenosine 26775-35-9 C18H22N6O4 386.41 —— N2,O5'-ditrityl-2-amino-6-chloro-9-[3-O-(tetrahydro-2-pyranyl)-β-D-arabinofuranosyl]purine 1053641-60-3 C53H48ClN5O5 870.448 —— 2-(2'-amino-6'-chloropurin-9'-yl)-5-ethylcarboxyamide-3,4-O-isopropylidene-tetrahydrofuran 936253-00-8 C15H19ClN6O4 382.807 —— 2-amino-N6-(m-methylbenzyl)adenosine —— C18H22N6O4 386.41 —— 2-amino-N6-[2''-(p-hydroxyphenyl)ethyl]adenosine 26884-84-4 C18H22N6O5 402.41 —— 2-amino-N6-amino-N6-methyladenosine —— C11H17N7O4 311.3 巴豆苷 isoguanosine 1818-71-9 C10H13N5O5 283.244 —— 2-amino-N6-(2-chloro-benzyl)-adenosine 26783-35-7 C17H19ClN6O4 406.829 —— N6-n-butyl-2-[(cyclohexylmethyl)amino]adenosine —— C21H34N6O4 434.539 2-氨基-N-(2-呋喃基甲基)腺苷 2-amino-N6-furfuryladenosine 26783-39-1 C15H18N6O5 362.345 —— 2-Amino-6-methoxy-9-(5'-O-tert-butyldimethylsilyl-β-D-ribofuranosyl)purine 132121-74-5 C17H29N5O5Si 411.533 O6-苄基鸟苷 6-O-benzylguanosine 4552-61-8 C17H19N5O5 373.368 —— (R,S)-N6-[Dihydroxypropyl]-2-amino-adenosine —— C13H20N6O6 356.338 —— (2R,3R,4R,5R)-4-(acetyloxy)-2-[(acetyloxy)methyl]-5-(6-chloro-2-cyano-9H-purin-9-yl)tetrahydro-3-furanyl acetate 333333-74-7 C17H16ClN5O7 437.796 —— N6-(2-phenylethyl)-2-<(phenylethyl)amino>adenosine 120524-43-8 C26H30N6O4 490.562 —— 2-amino-N6-(2-phenyl-propyl)-adenosine 26783-34-6 C19H24N6O4 400.437 —— 2-amino-N6-[2''-(m,p-dihydroxyphenyl)ethyl]adenosine —— C18H22N6O6 418.409 —— N6-(2''-phenylethyl)-2-[[2''-(p-hydroxyphenyl)ethyl]amino]adenosine —— C26H30N6O5 506.561 —— N6-[2''-(p-hydroxyphenyl)ethyl]-2-[[2''-(p-hydroxyphenyl)ethyl]amino]adenosine —— C26H30N6O6 522.561 —— N6-(2,2-diphenylethyl)-2-aminoadenosine 98383-43-8 C24H26N6O4 462.508 —— N6-tetrahydrofurfuryl-2-[(cyclohexylmethyl)amino]adenosine —— C22H34N6O5 462.549 —— 2-amino-N6-(2-methoxy-benzyl)-adenosine 26775-34-8 C18H22N6O5 402.41 —— 2-amino-6-[4-(pyrimidine-2-yl)piperazin-1-yl]-9-(β-D-ribofuranosyl)-9H-purine —— C18H23N9O4 429.439 —— (S)-O6-[2-Hydroxy-2-(p-methylphenyl)ethyl]guanosine 201403-22-7 C19H23N5O6 417.422 —— 2-amino-N6-[2''-(m-hydroxy-p-methoxyphenyl)ethyl]adenosine —— C19H24N6O6 432.436 —— 2-amino-N6-[2''-(p-hydroxy-m-methoxyphenyl)ethyl]adenosine —— C19H24N6O6 432.436 —— N6-(1-naphthylmethyl)-2-<(phenylmethyl)amino>adenosine 120524-46-1 C28H28N6O4 512.568 —— [(6aR,8R,9R,9aR)-8-(2-amino-6-chloropurin-9-yl)-2,2,4,4-tetra(propan-2-yl)-6a,8,9,9a-tetrahydro-6H-furo[3,2-f][1,3,5,2,4]trioxadisilocin-9-yl]oxy-phenoxymethanethione 170872-60-3 C29H42ClN5O6SSi2 680.372 —— 1,N2-ethenoguanosine 62462-38-8 C12H13N5O5 307.266 —— 9-[(2R,3R,5S)-3-[tert-butyl(dimethyl)silyl]oxy-5-[[tert-butyl(dimethyl)silyl]oxymethyl]oxolan-2-yl]-6-chloropurine-2-carbonitrile 132121-69-8 C23H38ClN5O3Si2 524.21 —— 2-amino-6-chloro-9-(2,3-dideoxy-β-D-glycero-pent-enofuranosyl)-9H-purine 141684-95-9 C10H10ClN5O2 267.675 —— 2-amino-6-chloro-9-[2-O-trifluoromethanesulfonyl-3,5-O-(1,1,3,3-tetraisopropyl-1,3-disiloxanyl)-β-D-ribofuranosyl]purine 675620-79-8 C23H37ClF3N5O7SSi2 676.261 —— isoguanosine-5'-O-[(phosphonomethyl)phosphonic acid] —— C11H17N5O10P2 441.231 2-碘-3'-脱氧腺苷 2-iodo-9-<3-deoxy-β-D-ribofuranosyl>adenine 115044-77-4 C10H12IN5O3 377.141 —— N6-(1''-naphthylmethyl)-2-[(cyclohexylmethyl)amino]adenosine —— C28H34N6O4 518.616 —— (2R,3S,4S,5R)-2-(2-cyano-6-methylamino-purin-9-yl)-5-hydroxy-methyl-tetrahydrofuran-3,4-diol 750644-43-0 C12H14N6O4 306.281 —— 2’,3’,5’-O-tri(t-butyldimethylsilyl)-O6-methylguanosine 869477-34-9 C29H57N5O5Si3 640.059 —— (5S,6R,8R9R)-6-(2-amino-6-methoxy-9H-purin-9-yl)-8-(hydroxymethyl)-1,7-dioxaspiro[4.4]nonan-9-ol 1378911-88-6 C14H19N5O5 337.335 —— (4S,5R,7R,8R)-5-(2-amino-6-methoxy-9H-purin-9-yl)-7-(hydroxymethyl)-1,6-dioxaspiro[3.4]octan-8-ol 1378911-92-2 C13H17N5O5 323.308 - 1
- 2
- 3
- 4
- 5
- 6
- 7
- 8
- 9
- 10
反应信息
-
作为反应物:描述:6-氯鸟嘌呤核苷 在 copper(l) iodide 、 氨 、 碘 、 亚硝酸异戊酯 作用下, 以 四氢呋喃 、 乙二醇甲醚 为溶剂, 反应 42.5h, 生成 (2R,3R,4S,5R)-2-[6-amino-2-(4-aminobutylamino)purin-9-yl]-5-(hydroxymethyl)oxolane-3,4-diol参考文献:名称:Adenosine Analogues as Inhibitors of Trypanosoma brucei Phosphoglycerate Kinase: Elucidation of a Novel Binding Mode for a 2-Amino-N6-Substituted Adenosine摘要:As part of a project aimed at structure-based design of adenosine analogues as drugs against African trypanosomiasis, N-6-, 2-amino-N-6-, and N-2-substituted adenosine analogues were synthesized and tested to establish structure-activity relationships for inhibiting Trypanosoma brucei glycosomal phosphoglycerate kinase (PGK), glyceraldehyde-3-phosphate dehydrogenase (GAPDH), and glycerol-3-phosphate dehydrogenase (GPDH). Evaluation of X-ray structures of parasite PGK, GAPDH, and GPDH complexed with their adenosyl-bearing substrates led us to generate a series of adenosine analogues which would target all three enzymes simultaneously. There was a modest preference by PGK for NG-substituted analogues bearing the 2-amino group. The best compound in this series, 2-amino-N-6-[2 "-(p-hydroxyphenyl)ethyl]adenosine (46b), displayed a 23-fold improvement over adenosine with an IC50 of 130 muM. 2-[[2 "-(p-Hydroxyphenyl)ethyl]amino]adenosine (46c) was a weak inhibitor of T. brucei PGK with an IC50 of 500 muM. To explore the potential of an additive effect that having the N-6 and N-2 substitutions in one molecule might provide, the best ligands from the two series were incorporated into N-6,N-2-disubstituted adenosine analogues to yield N-6-(2 " -phenylethyl)-2-[(2 " -phenylethyl)amino]adenosine (69) as a 30 muM inhibitor of T. brucei PGK which is 100-fold more potent than the adenosine template. In contrast, these series gave no compounds that inhibited parasitic GAPDH or GPDH more than 10-20% when tested at 1.0 mM. A 3.0 Angstrom X-ray structure of a T, brucei PGK/46b complex revealed a binding mode in which the nucleoside analogue was flipped and the ribosyl moiety adopted a syn conformation as compared with the previously determined binding mode of ADP. Molecular docking experiments using QXP and SAS program suites reproduced this "flipped and rotated" binding mode.DOI:10.1021/jm000287a
-
作为产物:参考文献:名称:新型6-取代氨基-9-(β -d-呋喃核糖基)嘌呤类似物的合成及其对人上皮癌细胞的生物活性摘要:制备了在C-6位置具有氮取代的新核苷衍生物,并通过NCI-磺胺丁丹B试验初步筛选了它们对人上皮癌细胞(肝Huh7,结肠HCT116,乳腺MCF7)的体外抗癌生物活性。N 6-(4-三氟甲基苯基)哌嗪类似物(27)表现出有希望的细胞毒性活性。 在Huh7,HCT116和MCF7细胞系上,化合物27比5-FU,氟达拉滨具有更高的细胞毒性(IC 50 = 1-4μM)。最有效的核苷(11,13,16,18,19,21,27,(28)进一步筛选了它们在肝细胞癌细胞系中的细胞毒性。化合物27对Huh7,Mahlavu和FOCUS细胞表现出最高的细胞毒活性(IC 50 分别为1、3和1μM)。分子的理化性质,药物相似性和药物得分曲线表明,它们被认为具有口服生物利用度。结果表明,新型衍生物将是潜在的药物候选物。DOI:10.1016/j.bmcl.2017.12.070
文献信息
-
[EN] 2'-SUBSTITUTED NUCLEOSIDE DERIVATIVES AND METHODS OF USE THEREOF FOR THE TREATMENT OF VIRAL DISEASES<br/>[FR] DÉRIVÉS DE NUCLÉOSIDE 2'-SUBSTITUÉS ET PROCÉDÉS D'UTILISATION DE CEUX-CI POUR LE TRAITEMENT DE MALADIES VIRALES申请人:MERCK SHARP & DOHME公开号:WO2012142085A1公开(公告)日:2012-10-18The present invention relates to 2'-Substituted Nucleoside Derivatives of Formula (I): and pharmaceutically acceptable salts thereof, wherein A, B, X, R1, R2 and R3 are as defined herein. The present invention also relates to compositions comprising at least one 2'-Substituted Nucleoside Derivative, and methods of using the 2'-Substituted Nucleoside Derivatives for treating or preventing HCV infection in a patient.
-
2'-AZIDO SUBSTITUTED NUCLEOSIDE DERIVATIVES AND METHODS OF USE THEREOF FOR THE TREATMENT OF VIRAL DISEASES申请人:Girijavallabhan Vinay公开号:US20140206640A1公开(公告)日:2014-07-24The present invention relates to 2′-Azido Substituted Nucleoside Derivatives of Formula (I): and pharmaceutically acceptable salts thereof, wherein B, X, R 1 , R 2 and R 3 are as defined herein. The present invention also relates to compositions comprising at least one 2′-Azido Substituted Nucleoside Derivative, and methods of using the 2′-Azido Substituted Nucleoside Derivatives for treating or preventing HCV infection in a patient.
-
[EN] CYCLIC DINUCLEOTIDES AS ANTICANCER AGENTS<br/>[FR] DINUCLÉOTIDES CYCLIQUES UTILISÉS EN TANT QU'AGENTS ANTICANCÉREUX申请人:BRISTOL MYERS SQUIBB CO公开号:WO2019074887A1公开(公告)日:2019-04-18The present invention is directed to compounds of the formula (I), wherein all substituents are defined herein, as well as pharmaceutically acceptable compositions comprising compounds of the invention and methods of using said compositions in the treatment of various disorders.本发明涉及式(I)的化合物,其中所有取代基在此定义,以及包括本发明化合物的药学上可接受的组合物,以及使用这些组合物治疗各种疾病的方法。
-
Nucleoside derivatives as inhibitors of RNA-dependent RNA viral polymerase申请人:——公开号:US20020147160A1公开(公告)日:2002-10-10The present invention provides nucleoside compounds and certain derivatives thereof which are inhibitors of RNA-dependent RNA viral polymerase. These compounds are inhibitors of RNA-dependent RNA viral replication and are useful for the treatment of RNA-dependent RNA viral infection. They are particularly useful as inhibitors of hepatitis C virus (HCV) NS5B polymerase, as inhibitors of HCV replication, and/or for the treatment of hepatitis C infection. The invention also describes pharmaceutical compositions containing such nucleoside compounds alone or in combination with other agents active against RNA-dependent RNA viral infection, in particular HCV infection. Also disclosed are methods of inhibiting RNA-dependent RNA polymerase, inhibiting RNA-dependent RNA viral replication, and/or treating RNA-dependent RNA viral infection with the nucleoside compounds of the present invention.
-
Pd/PTABS: Low-Temperature Thioetherification of Chloro(hetero)arenes作者:Siva Sankar Murthy Bandaru、Shatrughn Bhilare、Jesvita Cardozo、Nicolas Chrysochos、Carola Schulzke、Yogesh S. Sanghvi、Krishna Chaitanya Gunturu、Anant R. KapdiDOI:10.1021/acs.joc.9b00840日期:2019.7.19Heteroarenes of commercial relevance such as purines and pyrimidines were also found to be useful substrates for the reported transformation. The commercial drug Imuran (azathioprine) was synthesized as an example, and its preparation could be optimized. DFT studies were performed to understand the electronic effects of the tested ligands on the catalytic reaction.
表征谱图
-
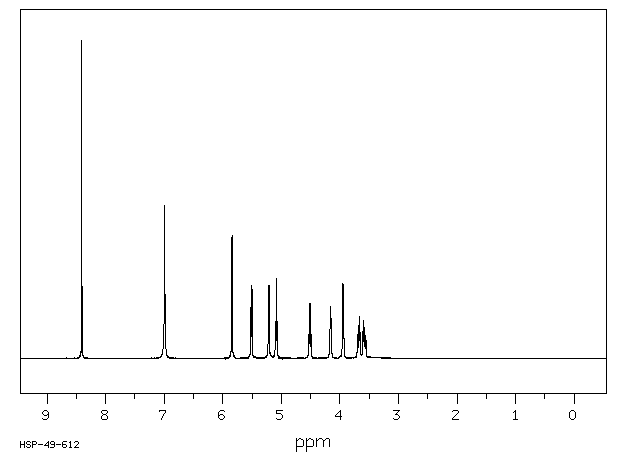
氢谱1HNMR
-
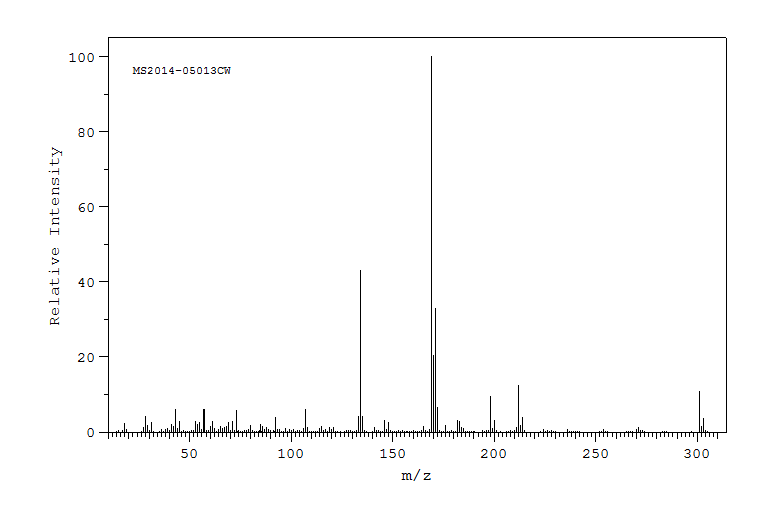
质谱MS
-
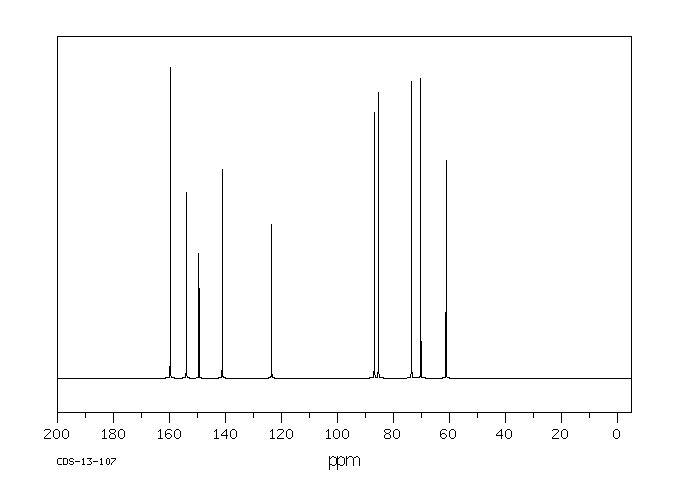
碳谱13CNMR
-
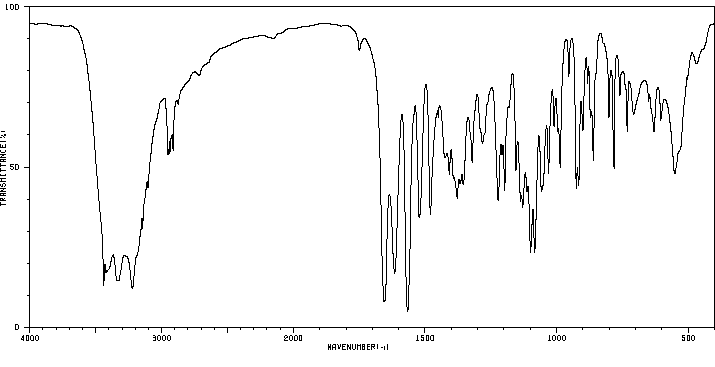
红外IR
-
拉曼Raman
-
峰位数据
-
峰位匹配
-
表征信息
同类化合物
黄质核苷
黄嘌呤核苷
鸟苷5'-硫代二磷酸酯
鸟苷,N,N-二甲基-6-O-[2-(4-硝基苯基)乙基]-
鸟苷 2',3',5'-三苯甲酸酯
鸟苷
马兜铃内酰胺A
顺式-玉米素-D-核糖甙
阿糖腺苷2',3',5'-三乙酸酯
阿糖腺苷 2',3'-二乙酸酯
阿糖腺苷
阿糖腺苷
阿糖肌苷
阿洛酮糖腺苷
阿斯卡霉素
虫草素
苯酰胺,N-[9-[2-脱氧-2-氟-3,5-二-O-(四氢-2H-吡喃-2-基)-β-D-呋喃阿拉伯糖基]-9H-嘌呤-6-基]-
苯酚,4-[4-(2-氨基乙基)-2-碘苯氧基]-,盐酸(1:1)
苏云金素
腺苷酸-5-羧酸
腺苷胺类
腺苷地尔
腺苷6-N-氨基磷酸酯
腺苷5-O-硫一磷酸二锂盐
腺苷5'-O-(1-硫代二磷酸酯)
腺苷-N(6)-二乙硫基醚-N'-吡啶氧杂亚胺5'-磷酸酯
腺苷-5'-羧酸2-氯乙酯
腺苷-5'-单烟酸酯
腺苷-5'-13C
腺苷-3(+2')-单磷酸单水合物
腺苷-2,8-T2
腺苷-2'-13C
腺苷-15NN1-氧化物
腺苷,8-(丁基氨基)-N-环戊基-
腺苷,2-溴-
腺苷,2-氯-N-环丙基-
腺苷,2-[(4-甲代戊基)氧代]-
腺苷,2-(苯基甲氧基)-
腺苷,2-(环己三烯并氧基)-
腺苷,2-(2-乙基丁氧基)-
腺苷,2',3'-O-(1-甲基亚乙基)-,5'-氨基磺酸酯
腺苷
肌苷肟
肌苷-8-14C
肌苷
美他卡韦
硒-(4-硝基苯甲酰基)-6-硒肌苷
甲基4,5-二溴-3-氟噻吩-2-羧酸酯
瑞加德松杂质3
瑞加德松杂质1
相关功能分类

联系我们



关注我们

公众号

