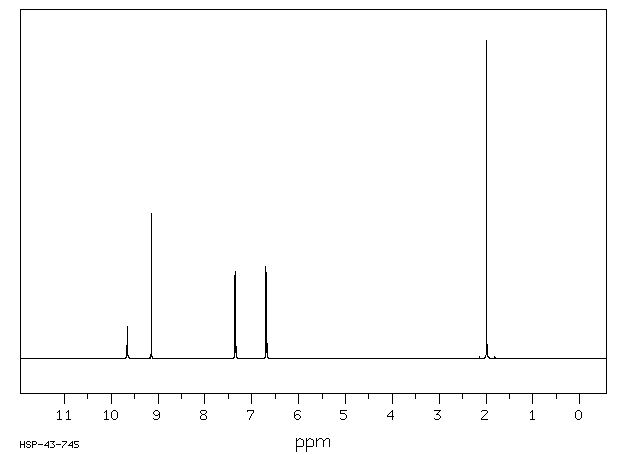
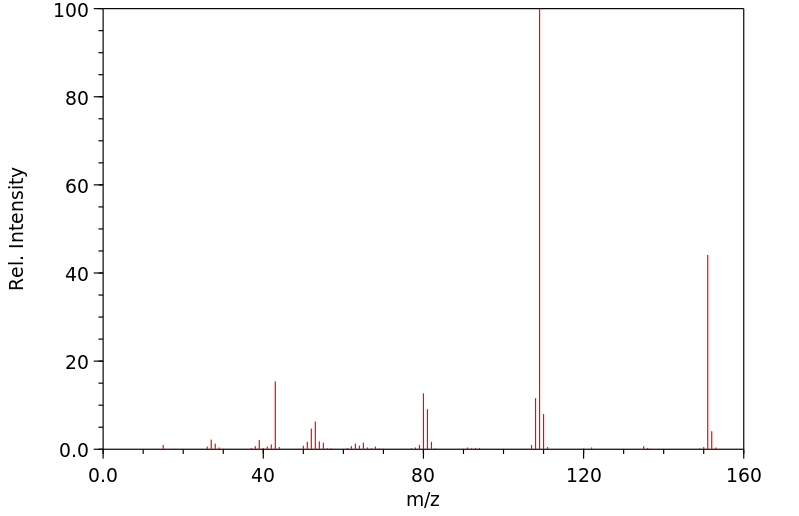
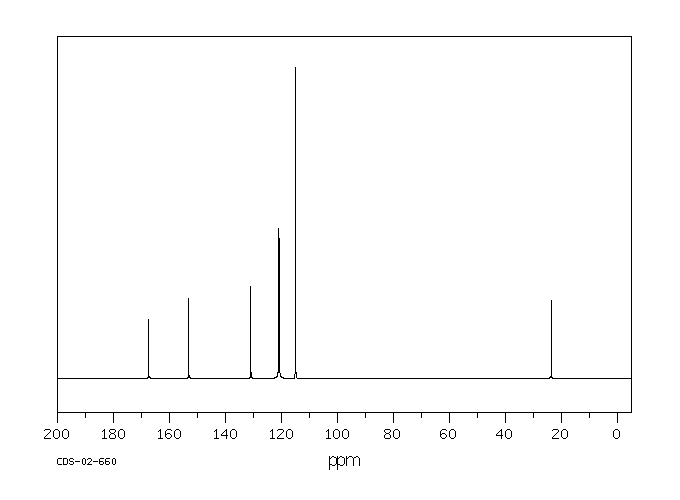
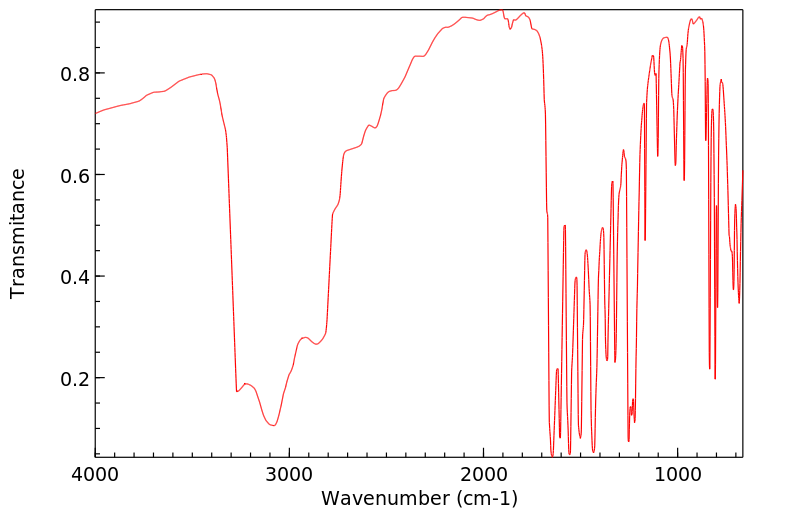
Acetaminophen is the major metabolite of _phenacetin_ and _acetanilid_. Acetaminophen is mainly metabolized in the liver by first-order kinetics and its metabolism of comprised of 3 pathways: conjugation with glucuronide, conjugation with sulfate, and oxidation through the cytochrome P450 enzyme pathway, mainly CYP2E1, to produce a reactive metabolite (N-acetyl-p-benzoquinone imine or NAPQI). At normal therapeutic doses, NAPQI undergoes fast conjugation with glutathione and is subsequently metabolized to produce both cysteine and mercapturic acid conjugates. High doses of acetaminophen (overdoses) can lead to hepatic necrosis due to the depletion of glutathione and of binding of high levels of reactive metabolite (NAPQI) to important parts of liver cells. The abovementioned damage to the liver can be prevented by the early administration of sulfhydryl compounds, for example, methionine and N-acetylcysteine.
About 80-85% of the acetaminophen in the body undergoes conjugation principally with glucuronic acid and to a lesser extent with sulfuric acid. Acetaminophen also is metabolized by microsomal enzyme systems in the liver.
In vitro and animal data indicate that small quantities of acetaminophen are metabolized by a cytochrome P-450 microsomal enzyme to a reactive intermediate metabolite (N-acetyl-p-benzoquinoneimine, N-acetylimidoquinone, NAPQI) which is further metabolized via conjugation with glutathione and ultimately excreted in urine as a mercapturic acid. It has been suggested that this intermediate metabolite is responsible for acetaminophen-induced liver necrosis and that high doses of acetaminophen may deplete glutathione so that inactivation of this toxic metabolite is decreased. At high doses, the capacity of metabolic pathways for conjugation with glucuronic acid and sulfuric acid may be exceeded, resulting in increased metabolism of acetaminophen by alternative pathways. In addition, it also has been suggested that in fasting individuals conjugation of high doses of acetaminophen with glucuronic acid may be reduced, secondary to decreased hepatic carbohydrate reserves and microsomal oxidation may be increased, resulting in increased risk of hepatotoxicity.
Yields 4-acetamidocatechol in rat; yields s-(5-acetamido-2-hydroxyphenyl)-l-cysteine probably in man. Yields p-acetamidophenyl-beta-d-glucuronide in rabbit; yields p-acetamidophenyl-beta-d-glucuronide in rat, in guinea pig, & in ferret; yields p-acetamidophenyl-beta-d-glucuronide in man & in dog; yields p-acetamidophenyl sulfate in rabbit, guinea pig, & ferret; yields p-acetamidophenyl sulfate in rat & in man; yields p-methoxyacetanilide in guinea pig; yields quinol probably in rat. /From table/
Children have less capacity for glucuronidation of the drug than do adults. A small proportion of acetaminophen undgoes n-hydroxylation to form n-acetyl-benzoquinoneimine, a highly reactive intermediate. This metabolite normally reacts with sulfhydryl groups in glutathione. However, after large doses of acetaminophen the metabolite is formed in amounts sufficient to deplete hepatic glutathione; under these circumstances reaction with sulfhydryl groups in hepatic proteins is increased and hepatic necrosis can result.
IDENTIFICATION AND USE: Acetaminophen is an odorless compound with a slightly bitter taste. It is a common analgesic and antipyretic agent used for the relief of fever as well as aches and pains associated with many conditions. HUMAN EXPOSURE AND TOXICITY: Nausea, vomiting, and abdominal pain usually occur within 2-3 hours after ingestion of toxic doses of the drug. In severe poisoning, CNS stimulation, excitement, and delirium may occur initially. This may be followed by CNS depression, stupor, hypothermia, marked prostration, rapid shallow breathing, rapid weak irregular pulse, low blood pressure, and circulatory failure. When an individual has ingested a toxic dose of acetaminophen, the individual should be hospitalized for several days of observation, even if there are no apparent ill effects, because maximum liver damage and/or cardiotoxic effects usually do not become apparent until 2-4 days after ingestion of the drug. Other symptoms of acute poisoning include cerebral edema and nonspecific myocardial depression. Vascular collapse results from the relative hypoxia and from a central depressant action that occurs only with massive doses. Shock may develop if vasodilation is marked. Fatal seizures may occur. Coma usually precedes death, which may occur suddenly or may be delayed for several days. Biopsy of the liver reveals centralobular necrosis with sparing of the periportal area. There have been reports of acute myocardial necrosis and pericarditis in individuals with acetaminophen poisoning. Hypoglycemia, which can progress to coma have been reported in patients ingesting toxic doses of acetaminophen. Low prothrombin levels and thrombocytopenia have been reported in patients with acetaminophen poisoning. Skin reactions of an erythematous or urticarial nature which may be accompanied by fever and oral mucosal lesions also have been reported. For use anytime during pregnancy, 781 exposures were recorded, and possible associations with congenital dislocation of the hip (eight cases) and clubfoot (six cases) were found. There is inadequate evidence in humans for the carcinogenicity of acetaminophen. ANIMAL TOXICITY STUDIES: There is inadequate evidence in experimental animals for the carcinogenicity of acetaminophen. In rats fasted 24 hours and given a single dose of acetaminophen (2 g/kg) by gavage, liver necrosis around the central vein was noted at 9-12 hours and was much more extensive at 24 hours after treatment. In mice after dietary exposure to acetaminophen up to 6400 mg/kg daily for 13 weeks hepatotoxicity, organ weight changes and deaths were observed. Cats are particularly susceptible to acetaminophen intoxication, developing more diffuse liver changes, while hepatic centrilobular lesions found in dogs. High doses of acetaminophen caused testicular atrophy and delay in spermiogenesis in mice. Furthermore, reductions in the fertility and neonatal survival in mice were seen in the F0 generation and decreases in F1 pup weights were found at acetaminophen dose 1430 mg/kg. Acetaminophen was not mutagenic in Salmonella typhimurium assay with or without metabolic activation in six strains: TA1535, TA1537, TA1538, TA100, TA97 and TA98. In vitro and animal data indicate that small quantities of acetaminophen are metabolized by a cytochrome P-450 microsomal enzyme to a reactive intermediate metabolite (N-acetyl-p-benzoquinoneimine, N-acetylimidoquinone, NAPQI) which is further metabolized via conjugation with glutathione and ultimately excreted in urine as a mercapturic acid. It has been suggested that this intermediate metabolite is responsible for acetaminophen-induced liver necrosis in cases of overdose. Excipients found in liquid formulations of acetaminophen may decrease its liver toxicity. ECOTOXICITY STUDIES: Daphnia magna was the most susceptible among the test organisms to the environmental effects of acetaminophen. Acetaminophen has recently been identified as a promising snake toxicant to reduce brown tree snake populations on Guam, while posing only the minimal risks to non-target rodents, cats, pigs and birds.
Paracetamol toxicity is one of the most common causes of poisoning worldwide. The toxic effects of acetaminophen are due to a minor alkylating metabolite (N-acetyl-p-benzo-quinone imine – also known as NAPQI), not acetaminophen itself nor any of the other major metabolites. Cytochromes P450 2E1 and 3A4 convert approximately 5% of paracetamol to NAPQI. This toxic metabolite reacts with sulfhydryl groups on proteins and with glutathione (GSH). NAPQI depletes the liver's natural antioxidant glutathione and directly damages cells in the liver, leading to liver failure. In animal studies, hepatic glutathione must be depleted to less than 70% of normal levels before hepatotoxicity occurs. More specifically, oxidation by NAPQI of GSH to GSSG (oxidized glutathione) and the reduction of GSSG back to GSH by the NADPH-dependent glutathione reductase appear to be responsible for the rapid oxidation of NADPH that occurs in hepatocytes incubated with NAPQI. Risk factors for toxicity include excessive chronic alcohol intake, fasting or anorexia nervosa, and the use of certain drugs such as isoniazid. At usual doses, paracetamol is quickly detoxified by combining irreversibly with the sulfhydryl group of glutathione to produce a non-toxic conjugate that is eventually excreted by the kidneys. The toxic dose of paracetamol is highly variable.
Chronic therapy with acetaminophen in doses of 4 grams daily has been found to lead to transient elevations in serum aminotransferase levels in a proportion of subjects, generally starting after 3 to 7 days, and with peak values rising above 3-fold elevated in 39% of persons. These elevations are generally asymptomatic and resolve rapidly with stopping therapy or reducing the dosage, and in some instances resolve even with continuation at full dose (Case 1).
While acetaminophen has few side effects when used in therapeutic doses, recent reports suggest that its standard use can result in severe hypersensitivity reactions including Stevens Johnson syndrome (SJS) and toxic epidermal necrolysis (TEN). Both of these syndromes can be life-threatening and both may be accompanied by evidence of liver injury. However, the hepatic involvement is usually mild and marked only by asymptomatic mild-to-moderate elevations in serum aminotransferase levels.
The best known form of hepatoxicity from acetaminophen is an acute, serious hepatocellular injury as a result of intentional or unintentional overdose. The injury is due to a direct, toxic effect of the high doses of acetaminophen. Acetaminophen hepatotoxicity most commonly arises after a suicide attempt using more than 7.5 grams (generally more than 15 grams) as a single overdose (Case 2). Hepatic injury generally starts 24 to 72 hours after the ingestion with marked elevations in serum ALT and AST (often to above 2000 U/L), followed at 48 to 96 hours by clinical symptoms: jaundice, confusion, hepatic failure and in some instances death. Evidence of renal insufficiency is also common. Serum aminotransferase levels fall promptly and recovery is rapid if the injury is not too severe. Similar injury can occur with high therapeutic or supratherapeutic doses of acetaminophen given over several days for treatment of pain and not as a purposeful suicidal overdose (Case 3). This form of acetaminophen hepatotoxicity is referred to as accidental or unintentional overdose, and usually occurs in patients who have been fasting, or are critically ill with a concurrent illness, alcoholism or malnutrition, or have preexisting chronic liver disease. Some cases of unintentional overdose occur in patients taking acetaminophen in combinations with controlled substances (oxycodone, codeine), who take more than recommended amounts over several days in attempts to control pain or withdrawal symptoms. Instances of unintentional overdose in children are often due to errors in calculating the correct dosage or use of adult sized tablets instead of child or infant formulations. Because acetaminophen is present in many products, both by prescription and over-the-counter, another problem occurs when a patient ingests full or high doses of several products unaware that several contain acetaminophen.
Likelihood score: A[HD] (well established cause of liver injury, but severe cases occur only with high doses).
Acetaminophen has 88% oral bioavailability and reaches its highest plasma concentration 90 minutes after ingestion. Peak blood levels of free acetaminophen are not reached until 3 hours after rectal administration of the suppository form of acetaminophen and the peak blood concentration is approximately 50% of the observed concentration after the ingestion of an equivalent oral dose (10-20 mcg/mL). The percentage of a systemically absorbed rectal dose of acetaminophen is inconsistent, demonstrated by major differences in the bioavailability of acetaminophen after a dose administered rectally. Higher rectal doses or an increased frequency of administration may be used to attain blood concentrations of acetaminophen similar to those attained after oral acetaminophen administration.
Acetaminophen metabolites are mainly excreted in the urine. Less than 5% is excreted in the urine as free (unconjugated) acetaminophen and at least 90% of the administered dose is excreted within 24 hours.
Volume of distribution is about 0.9L/kg. 10 to 20% of the drug is bound to red blood cells. Acetaminophen appears to be widely distributed throughout most body tissues except in fat.
Acetaminophen is rapidly and almost completely absorbed from the GI tract following oral administration. In healthy men, steady-state oral bioavailability of 1.3-g doses of extended-release tablets of acetaminophen administered every 8 hours for a total of 7 doses was equal to 1-g doses of conventional tablets of acetaminophen given every 6 hours for a total of 7 doses. Food may delay slightly absorption of extended-release tablets of acetaminophen. Following oral administration of immediate- or extended-release acetaminophen preparations, peak plasma concentrations are attained within 10-60 or 60-120 minutes, respectively. Following oral administration of a single 500-mg conventional tablet or a single 650-mg extended-release tablet, average plasma acetaminophen concentrations of 2.1 or 1.8 ug/mL, respectively, occur at 6 or 8 hours, respectively. In addition, dissolution of the extended-release tablets may depend slightly on the gastric or intestinal pH. Dissolution appears to be slightly faster in the alkaline pH of the intestines compared with the acidic pH of the stomach; however, this is of no clinical importance. Following administration of conventional preparations of acetaminophen, only small amounts of the drug are detectable in plasma after 8 hours. The extended-release tablets of acetaminophen release the drug for up to 8 hours, but in vitro data indicate that at least 95% of the dose is released within 5 hours.
[EN] S-NITROSOMERCAPTO COMPOUNDS AND RELATED DERIVATIVES<br/>[FR] COMPOSÉS DE S-NITROSOMERCAPTO ET DÉRIVÉS APPARENTÉS
申请人:GALLEON PHARMACEUTICALS INC
公开号:WO2009151744A1
公开(公告)日:2009-12-17
The present invention is directed to mercapto-based and S- nitrosomercapto-based SNO compounds and their derivatives, and their use in treating a lack of normal breathing control, including the treatment of apnea and hypoventilation associated with sleep, obesity, certain medicines and other medical conditions.
Compositions for Treatment of Cystic Fibrosis and Other Chronic Diseases
申请人:Vertex Pharmaceuticals Incorporated
公开号:US20150231142A1
公开(公告)日:2015-08-20
The present invention relates to pharmaceutical compositions comprising an inhibitor of epithelial sodium channel activity in combination with at least one ABC Transporter modulator compound of Formula A, Formula B, Formula C, or Formula D. The invention also relates to pharmaceutical formulations thereof, and to methods of using such compositions in the treatment of CFTR mediated diseases, particularly cystic fibrosis using the pharmaceutical combination compositions.
[EN] DIHYDROPYRROLONAPHTYRIDINONE COMPOUNDS AS INHIBITORS OF JAK<br/>[FR] COMPOSÉS DE DIHYDROPYRROLONAPHTYRIDINONE COMME INHIBITEURS DE JAK
申请人:TAKEDA PHARMACEUTICAL
公开号:WO2010144486A1
公开(公告)日:2010-12-16
Disclosed are JAK inhibitors of formula (I) where G1, R1, R2, R3, R4, R5, R6, and R7 are defined in the specification. Also disclosed are pharmaceutical compositions, kits and articles of manufacture which contain the compounds, methods and materials for making the compounds, and methods of using the compounds to treat diseases, disorders, and conditions involving the immune system and inflammation, including rheumatoid arthritis, hematological malignancies, epithelial cancers (i.e., carcinomas), and other diseases, disorders or conditions associated with JAK.
[EN] NOVEL COMPOUNDS AND PHARMACEUTICAL COMPOSITIONS THEREOF FOR THE TREATMENT OF INFLAMMATORY DISORDERS<br/>[FR] NOUVEAUX COMPOSÉS ET COMPOSITIONS PHARMACEUTIQUES LES COMPRENANT POUR LE TRAITEMENT DE TROUBLES INFLAMMATOIRES
申请人:GALAPAGOS NV
公开号:WO2017012647A1
公开(公告)日:2017-01-26
The present invention discloses compounds according to Formula (I), wherein R1, R3, R4, R5, L1, and Cy are as defined herein. The present invention also provides compounds, methods for the production of said compounds of the invention, pharmaceutical compositions comprising the same and their use in allergic or inflammatory conditions, autoimmune diseases, proliferative diseases, transplantation rejection, diseases involving impairment of cartilage turnover, congenital cartilage malformations, and/or diseases associated with hypersecretion of IL6 and/or interferons. The present invention also methods for the prevention and/or treatment of the aforementioned diseases by administering a compound of the invention.
[EN] COMPOUNDS AND THEIR USE AS BACE INHIBITORS<br/>[FR] COMPOSÉS ET LEUR UTILISATION EN TANT QU'INHIBITEURS DE BACE
申请人:ASTRAZENECA AB
公开号:WO2016055858A1
公开(公告)日:2016-04-14
The present application relates to compounds of formula (I), (la), or (lb) and their pharmaceutical compositions/preparations. This application further relates to methods of treating or preventing Αβ-related pathologies such as Down's syndrome, β- amyloid angiopathy such as but not limited to cerebral amyloid angiopathy or hereditary cerebral hemorrhage, disorders associated with cognitive impairment such as but not limited to MCI ("mild cognitive impairment"), Alzheimer's disease, memory loss, attention deficit symptoms associated with Alzheimer's disease, neurodegeneration associated with diseases such as Alzheimer's disease or dementia, including dementia of mixed vascular and degenerative origin, pre-senile dementia, senile dementia and dementia associated with Parkinson's disease.