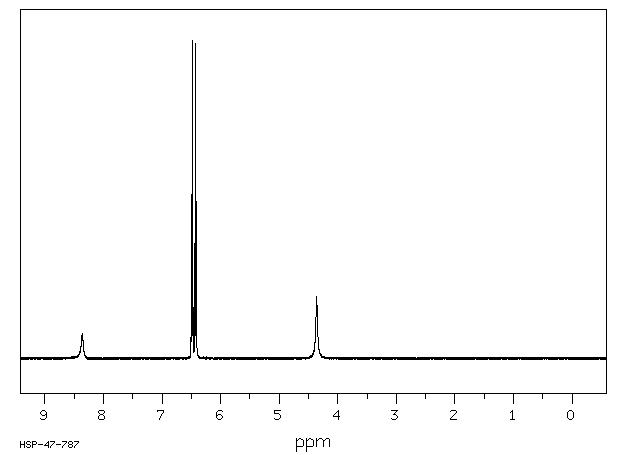
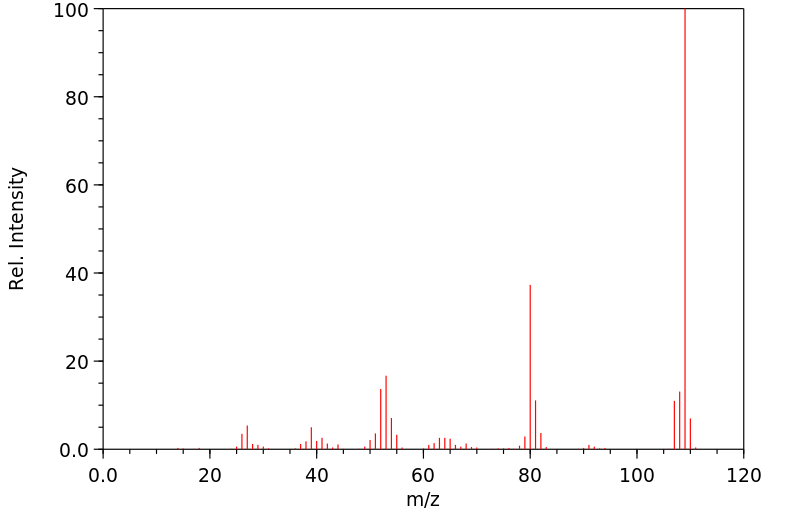
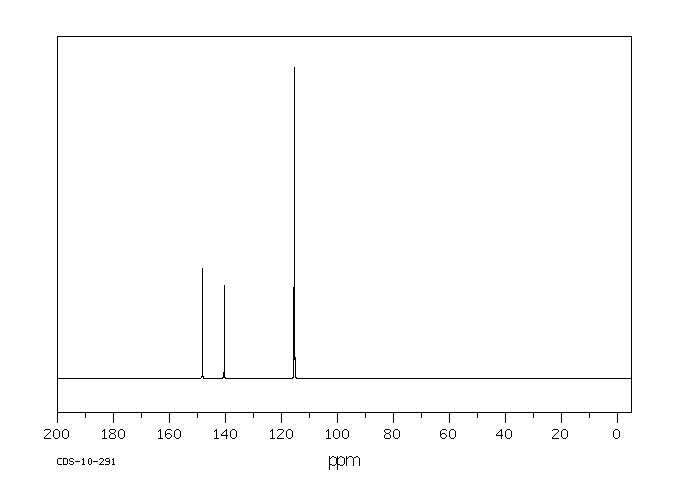
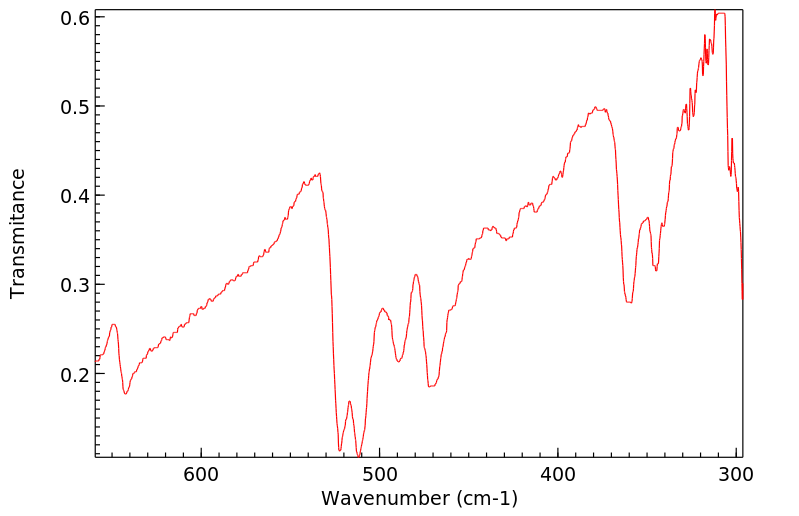
... Hepatocytes prepared from male Sprague-Dawley rats ... converted para-aminophenol (PAP) to two major metabolites (PAP-GSH conjugates and PAP-N-acetylcysteine conjugates) and several minor metabolites [PAP-O-glucuronide, acetaminophen (APAP), APAP-O-glucuronide, APAP-GSH conjugates, and 4-hydroxyformanilide]. Preincubating hepatoyctes with 1-aminobenzotriazole, an inhibitor of cytochromes P450, did not alter the pattern of PAP metabolism. In conclusion, we found that PAP was metabolized in hepatocytes predominantly to PAP-GSH conjugates and PAP-N-acetylcysteine conjugates in sufficient quantities to account for the nephrotoxicity of PAP.
... The hepatic metabolism of p-aminophenol in Wistar rats and the cytotoxicity of formed glutathione S-conjugates in rat renal epithelial cells /were examined/. After ip application of p-aminophenol (100 mg/kg), the following metabolites were identified in rat bile: 4-amino-2-(glutathion-S-yl)phenol, 4-amino-3-(glutathion-S-yl)-phenol, 4-amino-2,5-bis(glutathion-S-yl)phenol, 4-amino-2,3,5(or 6)-tris(glutathion-S-yl)phenol, an aminophenol conjugate (likely a sulfate or glucuronide), acetaminophen glucuronide, and 3-(glutathion-S-yl)acetaminophen. 4-Amino-3-(glutathion-S-yl)phenol, 4-amino-2,5-bis(glutathion-S-yl)phenol, and 4-amino-2,3,5(or 6)-tris(glutathion-S-yl)phenol induced a dose- and time-dependent loss of cell viability in rat kidney cortical cells. Cell killing was significantly reduced by inhibition of gamma-glutamyl transpeptidase with Acivicin. p-Aminophenol was also toxic to renal epithelial cells. Coincubation of p-aminophenol with tetraethylammonium bromide, a competitive inhibitor of the organic cation transporter, and with SKF-525A, an inhibitor of cytochrome P450, protected cells from p-aminophenol-induced toxicity. p-Aminophenol would thus be accumulated in the kidney mainly by organic cation transport systems, which are concentrated in the S-1 segment of the proximal tubule. However, p-aminophenol toxicity in vivo is directed toward the S-2 and S-3 segments, which are rich in gamma-glutamyl transpeptidase. These results and the observation that biliary cannulation and glutathione depletion reduce p-aminophenol nephrotoxicity suggest that the biosynthesis of toxic glutathione conjugates is responsible for p-aminophenol nephrotoxicity in vivo. The aminophenol glutathione S-conjugates formed induce p-aminophenol nephrotoxicity by a pathway dependent on gamma-glutamyl transpeptidase.
4-Aminophenol in the presence of oxyhemoglobin forms numerous adducts with glutathione (GSH). Using (14)C-4-aminophenol and (3)H-glutathione, ten different thioethers were isolated, by HPLC, with isotope ratios of 1:1, 1:2, 1:3, respectively ... In erythrocytes of humans and dogs, and in dog blood, in vivo, the same pattern of 4-aminophenol conjugates with GSH was found. In vivo, 5% of administered 4-aminophenol is converted into thioethers within erythrocytes, accompanied by a 60% decrease in the cellular GSH, indicating the role of erythrocytes in the biotransformation of xenobiotics.
4-Aminobiphenyl requires metabolic activation in order to exert its toxicity. This is catalyzed by N-hydroxylation via cytochrome P450 1A2, then followed by O-sulfation and O-acetylation by sulfotransferase 1A1 and arylamine N-acetyltransferase 2. The metabolites of 4-aminobiphenyl then form adducts with DNA, inducing mutations. 4-Aminobiphenyl and its metabolites may also cross the placenta and have fetal effects. (1, 2, 3, 4). It is also though that the mode of action involves metabolic activation by N-hydroxylation, followed by N-esterification leading to the formation of a reactive electrophile, which binds covalently to DNA, principally to deoxyguanosine, leading to an increased rate of DNA mutations and ultimately to the development of cancer. In humans and dogs, the urinary bladder urothelium is the target organ, whereas in mice it is the bladder and liver; in other species, other tissues can be involved. Differences in organ specificity are thought to be due to differences in metabolic activation versus inactivation (A15085).
4-Aminophenol may act as a skin sensitizer and cause contact dermatitis. In addition, inhalation of large amounts can cause methemoglobinemia and bronchial asthma. (T21)
4-Aminophenol may cause contact dermatitis. Signs and symptoms of methemoglobinemia may include shortness of breath, cyanosis, mental status changes, headache, fatigue, exercise intolerance, dizziness and loss of consciousness. Severe methemoglobinemia can result in dysrhythmias, seizures, coma, and death. (T21, L1613)
An absorption and excretion study of permanent hair dye intermediates containing 14-C was conducted in hairless Wistar rats under conditions of oxidation hair dyeing (ie, intermediates were mixed with H202 immediately before application). The cutaneous penetration of 14C-4-aminophenol (PAP) alone and in admixture with a nonradioactive coupler (3-amino-6-methylphenol, a 3-aminophenol (MAP) derivative) and that of the resultant 14C-indamine (N-[4-hydroxyphenyl}-3-amino-6-methyl benzoquinone imine) was determined. Hair dye solutions containing uniformly labeled PAP (0.75% or 70 nM) in a simple vehicle were applied to a 10 sq cm dorsal surface of up to 5 rats. Doses of PAP of 0.14 uM/sq cm, 0.69 uM/sq cm, and 3.44/ uM/sq cm yielded respective concentrations of 15.9 +/- 4.76 nM/sq cm, 52.04 +/- 6.73 nM/sq cm, and 58.4 +/- 11.5 nM/sq cm, which penetrated the skin and were detected in the excreta, the viscera, and the skin (excluding PAP found at the site of application) of the treated rats after 4 days. At the highest 14C-PAP concentration applied, the total quantity of 14C-PAP detected per sq cm of skin was approximately the same for PAP alone as for PAP mixed with nonradioactive MAP coupler (56.8 +/- 4.0 nM/sq cm). Penetration of the 14C-indamine was approximately 17 times less (3.6 +/- 0.46 nM) than that of PAP or of the mixture of PAP with the nonradioactive coupler..
Compositions for Treatment of Cystic Fibrosis and Other Chronic Diseases
申请人:Vertex Pharmaceuticals Incorporated
公开号:US20150231142A1
公开(公告)日:2015-08-20
The present invention relates to pharmaceutical compositions comprising an inhibitor of epithelial sodium channel activity in combination with at least one ABC Transporter modulator compound of Formula A, Formula B, Formula C, or Formula D. The invention also relates to pharmaceutical formulations thereof, and to methods of using such compositions in the treatment of CFTR mediated diseases, particularly cystic fibrosis using the pharmaceutical combination compositions.
Synthesis of a Novel Library of 1-Substituted Pyrido[1,2-a]benzimidazoles
作者:Satyanarayana Gadde、Yun Cheuk Leung、Mohan Bhadbade、Belamy B. Cheung、David StC. Black、Naresh Kumar
DOI:10.1071/ch20173
日期:——
The reactivity and synthesis of new analogues of pyrido[1,2-a]benzimidazoles have been explored. Twenty-three derivatives bearing phenoxy, thiophenoxy, aniline, and aryl groups at the 1-position were successfully synthesised in 25–91 % yield, via nucleophilic substitution, Buchwald–Hartwig amination, and Suzuki coupling type processes. Solvent free synthetic protocols were employed to achieve the nucleophilic
已经研究了吡啶并[1,2- a ]苯并咪唑的新类似物的反应性和合成。通过亲核取代,Buchwald-Hartwig胺化和Suzuki偶联类型工艺,成功合成了1位上带有苯氧基,噻吩氧基,苯胺和芳基的23种衍生物,收率25-91%。采用无溶剂的合成方案,通过空间需求中间体(7a)上的供电子基团或适度吸电子基团实现苯胺的亲核取代。在这项工作期间,一个不寻常的多环杂环被鉴定为副产物:二聚双(吡啶并[1,2- a ]苯并咪唑)。
Chemoselective Rearrangement Reactions of Sulfur Ylide Derived from Diazoquinones and Allyl/Propargyl Sulfides
作者:Sijia Yan、Junxin Rao、Cong-Ying Zhou
DOI:10.1021/acs.orglett.0c03493
日期:2020.11.20
three types of rearrangement reactions of sulfur ylidederivedfrom diazoquinones and allyl/propargyl sulfides. With Rh2(esp)2 as the catalyst, diazoquinones react with allyl/propargyl sulfides to form a sulfur ylide, which undergoes a chemoselective tautomerization/[2,3]-sigmatropic rearrangement reaction, a Doyle–Kirmse rearrangement/Cope rearrangement cascade reaction, or a Doyle–Kirmse rearrangement/elimination
[EN] AURORA KINASE MODULATORS AND METHOD OF USE<br/>[FR] MODULATEURS D'AURORA KINASE ET PROCÉDÉ D'UTILISATION
申请人:AMGEN INC
公开号:WO2009117157A1
公开(公告)日:2009-09-24
The present invention relates to chemical compounds having a general formula (I) wherein A1-5 and 7-8, D', L1, L2, R1, R3, R6-8, n and o are defined herein, and synthetic intermediates, which are capable of modulating the activity of Aurora kinase proteins and, thereby, influencing various disease states and conditions related to the activities of Aurora kinases. For example, the compounds are capable of influencing the process of cell cycle and cell proliferation to treat cancer and cancer-related diseases. The invention also includes pharmaceutical compositions, including the compounds, and methods of treating disease states related to the activity of Aurora kinase.