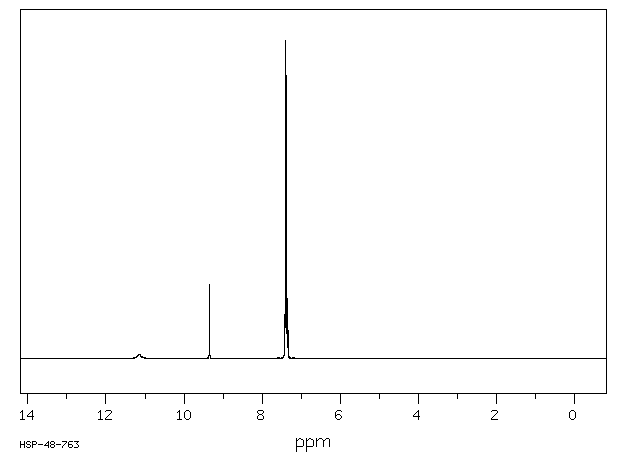
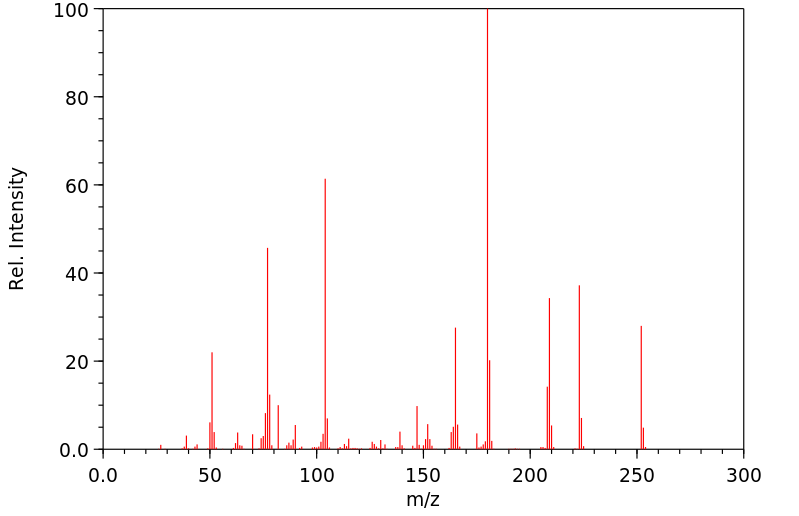
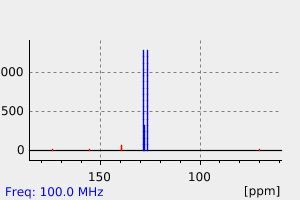
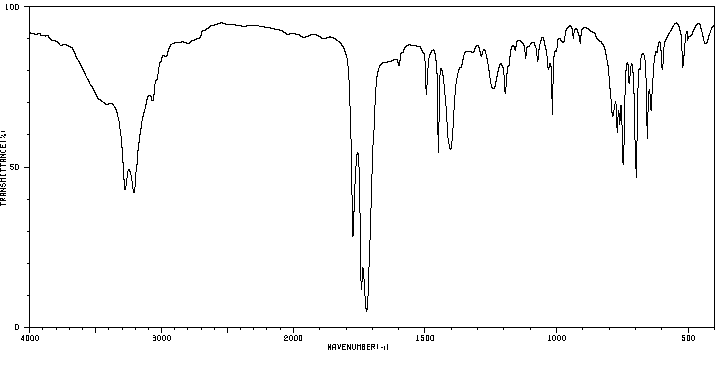
Phenytoin is extensively metabolized and is first transformed into a reactive _arene oxide intermediate_. It is thought that this reactive intermediate is responsible for many undesirable phenytoin adverse effects such as hepatotoxicity, SJS/TEN, and other idiosyncratic reactions. The _arene oxide_ is metabolized to either a _hydroxyphenytoin_ or _phenytoin dihydrodiol_ metabolite, although the former accounts for about 90% of phenytoin metabolism. Interestingly, two stereoisomers of the _hydroxyphenytoin_ metabolite are formed by CYP2C9 and CYP2C19: _(R)-p-HPPH_ and _(S)-p-HPPH_. When CYP2C19 catalyzes the reaction, the ratio of stereoisomers is roughly 1:1, whereas when CYP2C9 catalyzes the reaction, the ratio heavily favours the "S" stereoisomer. Since the metabolism of phenytoin is in part influenced by genetic polymorphisms of CYP2C9 and CYP2C19, this ratio can be utilized to identify different genomic variants of the enzymes. EPHX1, CYP1A2, CYP2A6, CYP2C19, CYP2C8, CYP2C9, CYP2D6, CYP2E1 and CYP3A4 are responsible for producing the _phenytoin dihydrodiol_ metabolite. _Hydroxyphenytoin_ can be metabolized by CYP2C19, CYP3A5, CYP2C9, CYP3A4, CYP3A7, CYP2B6 and CYP2D6 to a _phenytoin catechol_ metabolite or undergo glucuronidation by UGT1A6, UGT1A9, UGT1A1, and UGT1A4 to a _glucuronide metabolite_ that can be eliminated in the urine. On the other hand, the _phenytoin dihydrodiol_ entity is only transformed to the _catechol_ metabolite. The _catechol metabolite_ can undergo methylation by COMT and be subsequently eliminated in the urine, or can spontaneously oxidize to a _phenytoin quinone_ (NQO1 can transform the quinone back to the catechol metabolite). Of note, although CYP2C18 is poorly expressed in the liver, the enzyme is active in the skin and is involved in the primary and secondary hydroxylation of phenytoin. This CYP2C18 mediated bioactivation may be linked to the manifestation of adverse cutaneous drug reactions associated with phenytoin.
The major route of metabolism of phenytoin is oxidation by the liver to the inactive metabolite 5-(p-hydroxyphenyl)-5-phenylhydantoin (HPPH). Because this metabolism is a saturable process, small increases in dosage may produce substantial increases in plasma phenytoin concentrations...
The rate of hepatic biotransformation is increased in younger children, in pregnant women, in women during menses, and in patients with acute trauma; rate decreases with advancing age. The major inactive metabolite of phenytoin is 5-(p-hydroxyphenyl)-5-phenylhydantoin (HPPH). Phenytoin may be metabolized slowly in a small number of individuals due to genetic predisposition, which may cause limited enzyme availability and lack of induction.
... Oxidative metabolism of 1 of geminal phenyl rings of diphenylhydantoin ... 5-meta-hydroxyphenyl-(l) and 5-para-hydroxyphenyl-5-phenylhydantoin were detected in urine of man (approx ratio 1:12) ...
Phenytoin has known human metabolites that include 4-Hydroxyphenytoin, 3'-HPPH, (2S,3S,4S,5R)-6-(2,5-dioxo-4,4-diphenylimidazolidin-1-yl)-3,4,5-trihydroxyoxane-2-carboxylic acid, and 5-(3,4-dihydroxycyclohexa-1,5-dien-1-yl)-5-phenylimidazolidine-2,4-dione.
IDENTIFICATION: Phenytoin is an antiepileptic drug. Phenytoin is a white solid crystalline substance or a solid powder. It is odorless, and tasteless. It is soluble in water, alcohol and practically insoluble in chloroform, in ether and in methylene chloride. HUMAN EXPOSURE: Main risks and target organs: The intoxication usually manifests as mild central nervous system effects. More severe manifestations may be seen following massive overdose but fatalities are extremely rare. Summary of clinical effects: Onset of symptoms including lateral nystagmus, ataxia, and drowsiness occurs within 1 to 2 hours after ingestion and may persist for about 4 to 5 days. In more severe cases, horizontal nystagmus, coarse tremor and inability to walk may be observed. In very severe poisoning, conciousness is impaired but coma is rarely observed. Contraindications: Acute intermittent porphyria, hypersensitivity. Intravenous injection in patients with sino-atrial cardiac block, second- or third degree atrio-ventricular block, sinus bradycardia and Adam-Stokes syndrome. Caution is indicated in patients with uremia, hypoalbuminemia, liver function disorders and viral hepatitis. Routes of exposure: Oral: Most common route. Parenteral: Intravenously in status epilepticus. Absorption by route of exposure: Phenytoin is slowly, but almost completely absorbed from the gastro-intestinal tract; the rate of absorption is variable and its bioavailability can differ markedly with different pharmaceutical formulations. Large doses are more slowly absorbed. In severe oral poisoning, gastro-intestinal absorption may continue up to 60 hours. Administration of 100 mg orally to normal volunteers produced two peaks at 2.5 to 3.5 and 10 to 12 hours. Distribution by route of exposure: Phenytoin is widely distributed throughout the body and is extensively (87 to 93%) bound to protein. Plasma binding is almost exclusively to albumin; in individuals with normal plasma albumin concentration and in absence of displacing agents, phenytoin is about 90% plasma bound. Biological half-life by route of exposure: Following oral administration of therapeutic doses, phenytoin has a very variable, dose-dependent half-life. The range for a therapeutic dose is from 8 to 60 hours with an average of from 20 to 30 hours. In overdose in adults the range is from 24 to 230 hours. Metabolism: Phenytoin is extensively metabolizd in the liver to 5-(4-hydroxyphenyl)-5 phenyl-hydantoin, which is inactive. This para hydroxylation of phenytoin is carried out by cytochrome P450 2C9. This enzyme also hydroxylates tolbutamide, and warfarin. This explains the interaction with these substances. The p-hydroxylated phenytoin is in turn conjugated to its glucuronide. Phenytoin hydroxylation is capacity limited because of the saturable enzyme systems in the liver. At therapeutic doses, metabolism is nonlinear (first order kinetics), while at toxic doses the metabolism is linear (zero order kinetics). The p-hydroxylated phenytoin can be oxidized to 3,4- dihydroxyphenyl-phenylhydantoin, the catechol metabolite of phenytoin, and further to the 3-O-methylated catechol metabolite of phenytoin. These metabolites of phenytoin are of possible toxicological interest. Phenytoin is more rapidly metabolised in children. The rate of metabolizm appears to be subject to genetic polymorphism. Phenytoin undergoes entero-hepatic recycling. Elimination by route of exposure: Phenytoin is mainly excreted in the urine as its hydroxylated metabolite (23 to 70%), either free or in conjugated form (5%). About 4% is excreted unchanged, in the urine and 5% in the feces. Small amounts are excreted in the milk. Mode of action: Toxicodynamics: Phenytoin is eliminated mainly through para-hydroxylation by a cytochrome P450 system. The metabolic pathway is subject to saturable kinetics in overdose, allowing accumulation of free phenytoin. Even at therapeutic doses, accumulation of free phenytoin is possible in: hypoalbuminemia, chronic renal failure, hepatic dysfunction, hereditary insufficient para-hydroxylation, and inhibition of phenytoin metabolism by other drugs. Pharmacodynamics: Phenytoin binds to specific site on voltage- dependent sodium channels and is thought to exert its anticonvulsant effect by suppressing the sustained repetitive firing of neurons by inhibiting sodium flux through these voltage dependent channels. Phenytoin stabilizes membranes, protecting the sodium pump in the brain and in the heart. It limits the development of maximal convulsive activity and reduces the spread of convulsive activity from a discharging focus without influencing the focus itself. Phenytoin has antiarrhythmic properties similar to those of quinidine or procainamide. Although phenytoin has minimal effect on the electrical excitability of cardiac muscle, it decreases the force of contraction, depresses pacemaker action and improves atrioventricular conduction. It also prolongs the effective refractory period relative to the action potential duration. Carcinogenicity: Malignancies, including neuroblastoma, in children whose mothers were on phenytoin during pregnancy have been reported. Teratogenicity: Phenytoin is classed as a teratogen risk factor D (Positive evidence of human fetal risk, but the benefits from use in pregnant women may be acceptable despite the risk). The epileptic pregnant woman taking phenytoin, either alone or in combination with other anticonvulsants, has a two to three times greater risk of delivering a child with congenital defects. It is not known if this increased risk is due to antiepileptic drugs, the disease itself, genetic factors, or a combination of these, although some evidence indicates that drugs are the causative factor. A recognizable pattern of malformations, known as the fetal hydantoin syndrome has been described and includes craniofacial and limb abnormalities, cleft lip, impaired growth, and congenital heart defects. Drugs displacing phenytoin plasma protein binding sites: Azapropazone, diazoxide, heparin, ibuprofen, phenylbutazone, salicylic acid, sulfadimethoxine, sulfafurazole, sulfamethizole, sulfamethoxydiazine, sulfamethoxypyridazine, tolbutamide and valproic acid. Decreased total and unbound plasma phenytoin concentration: caused by increased metabolism: Folic acid, Dexamethasone, Phenobarbital, Diazepam, Rifampicin, Methadone, Nitrofurantoin, Estrogens and progestagens. Increased unbound fraction of phenytoin secondary to reduced: intrinsic metabolism: Anticonvulsants: Valproic acid, Carbamazepine, Sulthiame, Clobazam, Antithrombotics: Coumarin derivatives and Triclodine, Antituberculous drugs: Isoniazid and PAS, H2 antagonists: Cimetidine, Ranitidine and Omeprazole. Non-steroidal anti-inflammatory agents: Azapropazone, Phenylbutazone, Ibuprofen.Antiinfective agents: Metronidazole and Chloramphenicol Antimycotics: Miconazole, Fluconazole. Psychoactive drugs: Fluoxetine, Risperidone . Miscellaneous: Amiodarone, Allopurinol and Disulfiram. Main adverse effects: Anticonvulsant hypersensitivity syndrome is a potentially fatal drug reaction with cutaneous and systemic manifestations. The findings are: fever, dermatological: erythema, papulous rash. In some cases erythroderma and even a lethal epidermal necrolysis has been reported. Lymphadenopathy (70%): lymphoma and depressed immunological function have been reported. Hepatitis: hepatitis may develop with severe liver failure and death. Hematological: leukocytosis with atypical lymphocytes, eosinophilia, and agranulocytosis. Connective tissues: coarsening of facial features, enlargement of the lips, gingival hyperplasia, hypertrichosis, Peyronie's disease. Clinical effects: Acute poisoning: Ingestion: Onset of symptoms and signs, principally involving the central nervous system, occurs within hours of acute overdose. These manifestations of toxicity may last many days and, in general, correlate with serum phenytoin concentrations. The earliest manifestations of toxicity following overdose are nystagmus on lateral gaze, ataxia and drowsiness. With more severe intoxication, vertical nystagmus, dysarthria, progressive ataxia to the point of inability to walk, hyperreflexia and impaired level of consciousness are observed. Coma and/or respiratory depression is rarely observed and should prompt consideration of an alternative diagnosis. Paradoxical seizures have been reported in severe phenytoin intoxication but are extremely rare. Parenteral exposure: Fatalities have been reported following intravenous administration of phenytoin to elderly patients with cardiac arrhythmias. These complications appear more likely when intravenous phenytoin is administered at a rapid rate and have been attributed to the solvent propylene glycol rather than to the phenytoin itself. The risk of hypotension and arrythmia is minimal when intravenous phenytoin is used as an anticonvulsant and administered at the recommended rate. In a series of 164 patients who received intravenous phenytoin loading following presentation with acute convulsions, the incidence of hypotension was approximately 5%, and the incidence of apnea and cardiac arrhythmias was 0%. Course, prognosis, cause of death: Normally the clinical course is one of gradual resolution of the signs and symptoms of intoxication leading to complete recovery. Death is rare after phenytoin overdose. It has been reported in association with administration of intravenous phenytoin for treatment of cardiac arrhythmias in elderly people, and rarely from coma and hypotension following oral overdose in children. Systematic description of clinical effects: Cardiovascular: Intravenous phenytoin has been reported to cause depression of cardiac conduction, ventricular fibrillation and heart block in elderly people treated for cardiac arrhythmias. Intravenous phenytoin is irritant and may cause phlebitis. Neurological: CNS: Nystagmus, ataxia, dysarthria, drowsiness, coarse resting tremor, ankle clonus, brisk deep tendon reflexes. In severe poisoning, the patient becomes obtunded, confused and disoriented. Coma and respiratory depression are unusual. Hepatic: A phenytoin hypersensitivity syndrome occurs and is characterised by hepatitis. Overall mortality rate when liver is involved is between 18% and 40%. The hepatitis is usually anicteric. Icterus indicates a poorer prognosis. Hepatomegaly with or without plenomegaly may be present. The elevated hepatic transaminases, which may be in the thousands of international units, can continue to rise after phenytoin is discontinued. Phenytoin induced chronic hepatitis has been reported. Phenytoin can induce hyperglycemia by inhibiting the release of insulin. However, hypoglycemia has been reported in a patient treated with phenytoin for 19 years who ingested 20 g phenytoin together with 225 mg zopiclone. Dermatological: In the anticonvulsant hypersensitivity syndrome, the cutaneous eruption begins as a patchy macular erythema that evolves into a dusky, pink red, confluent, papular rash that usually is pruritic. The upper trunk, face, and upper extremities are affected first, with later involvement of the lower extremities. In some cases erythroderma ensues. Patients have periorbital and facial edema. Epidermal necrolysis (even lethal) has been reported. Hematological: A number of adverse haematological effects have been reported. These are not observed following acute overdose. The hematological abnormalities reported include leucocytosis with atypical lymphocytes, eosinophilia, leucopenia and agranulocytosis. The marrow toxicity of anticonvulsants, which may be more likely when used in combination (primidone), is recognized. There may be three mechanisms of toxicity. Firstly, primidone and phenytoin both cause folate deficiency and a megaloblastic anemia. Secondly, an immune mechanism with a phenytoin-dependent antigranulocyte antibody may cause leucopenia, which resolves on discontinuing therapy. Finally, phenytoin may cause a direct toxic effect with pancytopenia and agranulocytosis. Subnormal serum folate concentrations were found in patients with chronic epilepsy treated with phenytoin. It was suggested that folate deficiency resulted from accelerated metabolism of folate consequent upon induction of liver enzymes by anticonvulsants. Immunological: It seems likely that an etiological relationship exists between phenytoin treatment and lymphoma. There is evidence of depressed immunological function in patients given phenytoin. Allergic reactions: Anticonvulsant hypersensitivity syndrome is a potentially fatal drug reaction with cutaneous and systemic manifestations to the arene oxide producing anticonvulsants: phenytoin, carbamazepine, and phenobarbital sodium. The features include fever, rash, lympheadenopathy, periorbital or facial edema, hepatitis, hematologic abnormalities, myalgia, arthralgia and pharyngitis. The reaction may be genetically determined. Other: Connective tissues: coarsening of facial features, enlargment of the lips, gingival hyperplasia and hypertrichosis. /Phenytoin/
Phenytoin acts on sodium channels on the neuronal cell membrane, limiting the spread of seizure activity and reducing seizure propagation. By promoting sodium efflux from neurons, phenytoin tends to stabilize the threshold against hyperexcitability caused by excessive stimulation or environmental changes capable of reducing membrane sodium gradient. This includes the reduction of post-tetanic potentiation at synapses. Loss of post-tetanic potentiation prevents cortical seizure foci from detonating adjacent cortical areas.
Prospective studies indicate that a fairly high proportion of patients taking phenytoin have transient serum aminotransferase elevations. These elevations are usually benign, not associated with liver histological abnormalities and usually resolve even with drug continuation. In addition, a higher proportion of patients have mild-to-moderate elevations in gammaglutamyl transpeptidase (GGT) levels, which is indicative of hepatic enzyme induction rather than liver injury. Marked aminotransferase elevations (>3 fold elevated) occur rarely.
Importantly, however, phenytoin is one of the most common causes of clinically apparent drug induced liver disease and acute liver failure. More than 100 cases of liver injury due to phenytoin (diphenylhydantoin) have been published and a characteristic clinical pattern (signature) of injury has been described. The estimated frequency ranges from 1 per 1000 to 1 per 10,000 and probably varies by race and ethnicity. The typical case arises after 2 to 8 weeks of therapy with initial onset of fever, rash, facial edema and lymphadenopathy, followed in a few days by jaundice and dark urine. The serum enzyme elevations can be hepatocellular, although mixed patterns are probably more common and rare cases are cholestatic. Eosinophilia, increased white counts and atypical lymphocytosis are also common. Autoantibody formation is rare. The clinical symptoms and signs can mimic acute mononucleosis or even lymphoma (pseudo-lymphoma syndrome). Almost all cases of phenytoin hepatotoxicity occur in the context of a systemic hypersensitivity syndrome and it is referred to often as the anticonvulsant hypersensitivity syndrome (HDS) or drug rash with eosinophilia and systemic symptoms syndrome (DRESS). Other manifestations can be Stevens-Johnson syndrome, toxic epidermal necrolysis, aplastic anemia, thrombocytopenia, neutropenia, nephritis, and pneumonitis. Most cases of liver injury are self-limiting and resolve within 1 to 2 months of stopping phenytoin. However, the liver injury can be severe and many fatal instances have been reported, phenytoin usually appearing in the top 10 causes of drug induced acute liver failure. In the typical case, however, recovery is usually complete.
Likelihood score: A (well known cause of clinically apparent liver injury).
Given its narrow therapeutic index, therapeutic drug monitoring is recommended to help guide dosing. Phenytoin is completely absorbed. Peak plasma concentration is attained approximately 1.5-3 hours, and 4-12 hours after administration of the immediate release formulation and the extended release formulation, respectively. It should be noted that absorption can be markedly prolonged in situations of acute ingestion.
The clearance of phenytoin is non-linear. At lower serum concentrations (less than 10 mg/L), elimination is characterized by first order kinetics. As plasma concentrations increase, the kinetics shift gradually towards zero-order, and finally reach zero-order kinetics once the system is saturated.
Studies using Dilantin have shown that phenytoin and its sodium salt are usually completely absorbed from the GI tract. Bioavailability may vary enough among oral phenytoin sodium preparations of different manufacturers to result in toxic serum concentrations or a loss of seizure control (subtherapeutic serum concentrations)...
Palladium Catalyzed C-Arylation of Amino Acid Derived Hydantoins
摘要:
Palladium(II) trifluoroacetate (5 mol %) catalyzes the C-arylation of N,N-disubstituted hydantoins by aryl iodides in good yield. The reaction proceeds through base-promoted enolization of the amino acid derived hydantoins, and the resulting 5,5-disubstituted hydantoins may be deprotected at one or both N atoms to yield biologically active structures or alternatively hydrolyzed to the parent a-aryl a-amino acids. The reaction is successful with a variety of parent amino acids and a range of electron-rich and electron-poor aryl iodides.
[EN] ACC INHIBITORS AND USES THEREOF<br/>[FR] INHIBITEURS DE L'ACC ET UTILISATIONS ASSOCIÉES
申请人:GILEAD APOLLO LLC
公开号:WO2017075056A1
公开(公告)日:2017-05-04
The present invention provides compounds I and II useful as inhibitors of Acetyl CoA Carboxylase (ACC), compositions thereof, and methods of using the same.
[EN] COMPOUNDS AND THEIR USE AS BACE INHIBITORS<br/>[FR] COMPOSÉS ET LEUR UTILISATION EN TANT QU'INHIBITEURS DE BACE
申请人:ASTRAZENECA AB
公开号:WO2016055858A1
公开(公告)日:2016-04-14
The present application relates to compounds of formula (I), (la), or (lb) and their pharmaceutical compositions/preparations. This application further relates to methods of treating or preventing Αβ-related pathologies such as Down's syndrome, β- amyloid angiopathy such as but not limited to cerebral amyloid angiopathy or hereditary cerebral hemorrhage, disorders associated with cognitive impairment such as but not limited to MCI ("mild cognitive impairment"), Alzheimer's disease, memory loss, attention deficit symptoms associated with Alzheimer's disease, neurodegeneration associated with diseases such as Alzheimer's disease or dementia, including dementia of mixed vascular and degenerative origin, pre-senile dementia, senile dementia and dementia associated with Parkinson's disease.
4' SUBSTITUTED COMPOUNDS HAVING 5-HT6 RECEPTOR AFFINITY
申请人:Dunn Robert
公开号:US20080318941A1
公开(公告)日:2008-12-25
The present disclosure provides compounds having affinity for the 5-HT
6
receptor which are of the formula (I):
wherein R
1
, R
2
, R
5
, R
6
, B, D, E, G, Q, x and n are as defined herein. The disclosure also relates to methods of preparing such compounds, compositions containing such compounds, and methods of use thereof.
Heterobicyclic compounds of Formula (I):
or a pharmaceutically-acceptable salt, tautomer, or stereoisomer thereof, as defined in the specification, and compositions containing them, and processes for preparing such compounds. Provided herein also are methods of treating disorders or diseases treatable by inhibition of PDE10, such as obesity, non-insulin dependent diabetes, schizophrenia, bipolar disorder, obsessive-compulsive disorder, Huntington's Disease, and the like.
Formula (I)的杂环化合物:
或其药用可接受的盐、互变异构体或立体异构体,如规范中所定义,并含有它们的组合物,以及制备这种化合物的方法。本文还提供了通过抑制PDE10来治疗由此可治疗的疾病或疾病的方法,如肥胖症、非胰岛素依赖型糖尿病、精神分裂症、躁郁症、强迫症、亨廷顿病等。