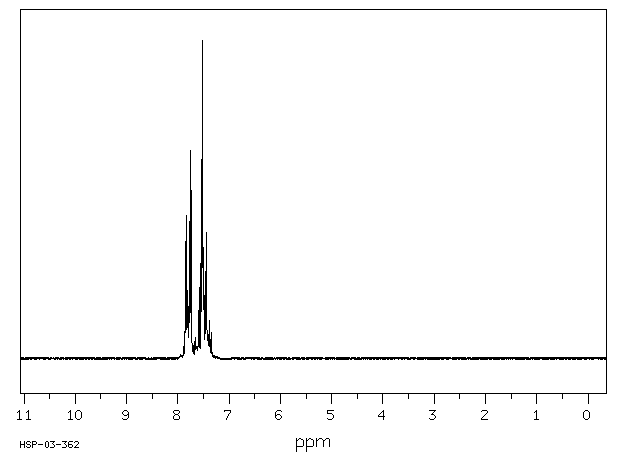
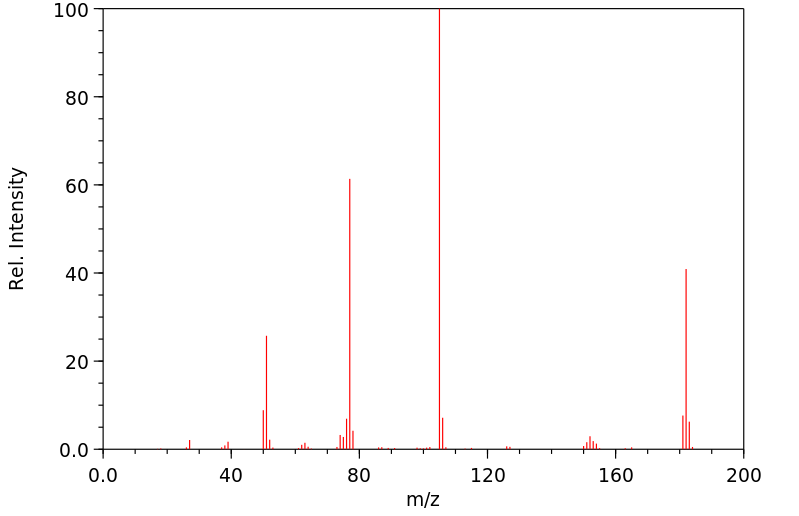
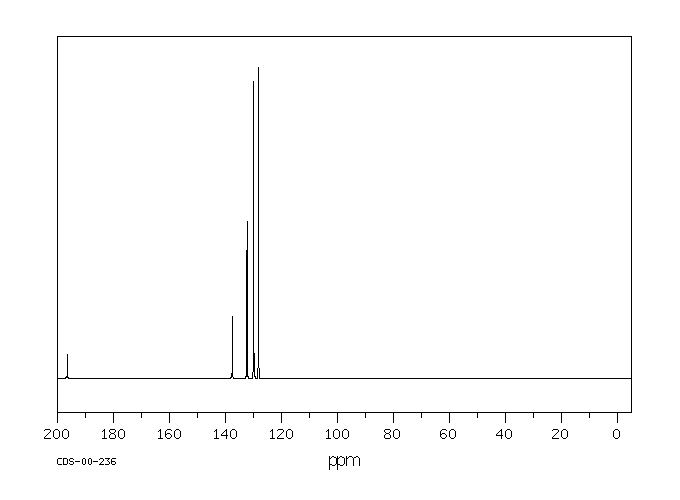
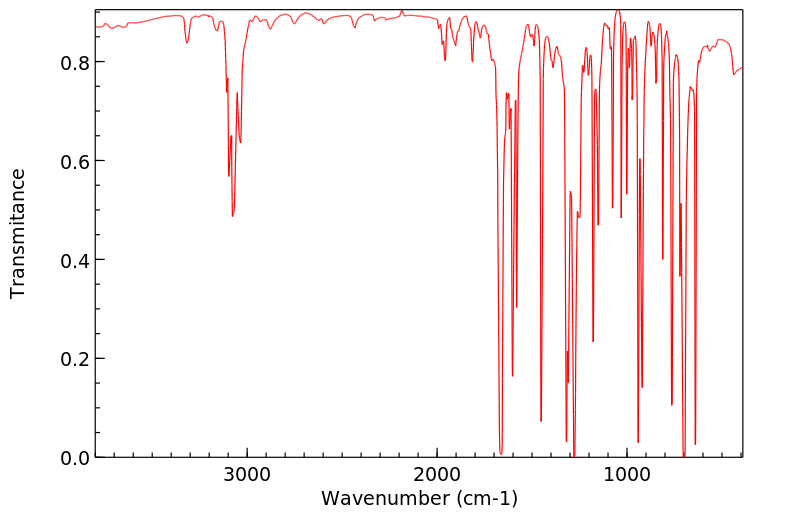
In male Sprague-Dawley rats that received benzophenone by gavage, 1% of the administered dose was detected as p-hydroxybenzophenone in enzymetreated urine samples, but not in unhydrolyzed urine (Stocklinski et al., 1979). No p-hydroxybenzophenone was detected in the feces.
The results from recent studies show that some benzophenones (BPs) and their hydroxylated metabolites can function as weak estrogens (E2) in the environment. However, little is known about the structure-activity relationship of these molecules. We have examined the effects of exposure to ten different BPs on the proliferation of estrogen receptor (ER)-positive breast cancer cells and on the transcriptional activity of E2-target genes. We analyzed two genes that are tightly linked with estrogen-mediated proliferation, the CXCL12 and amphiregulin genes and two classical estrogen-responsive genes, the pS2 and progesterone receptor. Significant differences in the BPs efficiency to induce cell proliferation and endogenous E2-target gene expressions were observed. Using ERE-, Sp1-, AP1- and C3-reporter genes that contain different ER-binding sites in their promoter, we also showed significant differences in the BPs efficiency in activation of the ER transactivation. Together, our analyzes showed that the most active molecule is 4-hydroxy-BP. Docking analysis of the interaction of BPs in the ligand-binding pocket of ERa suggests that the minimum structural requirement for the estrogenic activity of BPs is a hydroxyl (OH) group in the phenyl A-ring that allows interaction with Glu-353, Arg-394 or Phe-404, which enhances the stability between BPs and ERa. Our modeling also indicates a loss of interaction between the OH groups of the phenyl B-ring and His-524. In addition, the presence of some OH groups in the phenyl B-ring can create repulsion forces, which may constrain helix 12 in an unfavorable position, explaining the differential estrogenic effects of BPs. These results, together with our analysis of BPs for their potency in activation of cell proliferation and ER-mediated transcription, report an improved understanding of the mechanism and structure-activity relationship of BPs. /benzophenones/
Benzophenone's main metabolic pathway in the rabbit is by reduction to benzhydrol, which is excreted in urine conjugated with glucoronic acid. Small amount (1%) is converted to p-hydroxybenzophenone following oral administration to rats. No p-hydroxybenzohydrol was detected in urine or feces.
来源:Hazardous Substances Data Bank (HSDB)
代谢
苯并苯酮是西那rizine的人类已知代谢物。
Benzophenone is a known human metabolite of cinnarizine.
Paraoxonase (PON1) is a key enzyme in the metabolism of organophosphates. PON1 can inactivate some organophosphates through hydrolysis. PON1 hydrolyzes the active metabolites in several organophosphates insecticides as well as, nerve agents such as soman, sarin, and VX. The presence of PON1 polymorphisms causes there to be different enzyme levels and catalytic efficiency of this esterase, which in turn suggests that different individuals may be more susceptible to the toxic effect of OP exposure.
IDENTIFICATION AND USE: Benzophenone (BZP) comes in the form of orthorhombic prisms, monoclinic prisms, and white crystals. It has a characteristic sweet geranium or rose-like odor. BZP is used as a photoinitiator, a fragrance enhancer, an ultraviolet stabilizer, and, occasionally, as a flavor ingredient; it is also used in the manufacture of insecticides, agricultural chemicals, pharmaceuticals (antihistamines), hypnotics, paints, lacquers and cosmetics; and is an additive for plastics, coatings, and adhesives. Not registered for current use as a pesticide in the U.S., but approved pesticide uses may change periodically and so federal, state and local authorities must be consulted for currently approved uses. HUMAN EXPOSURE AND TOXICITY: There is limited information on the toxic effects of BZP in humans published. It showed little activity in the hormone-responsive reporter assay tested in various cell lines. ANIMAL STUDIES: BZP showed no estrogenic activity, however, after irradiating an aqueous solution of BZP with UV or sunlight, it can be converted into ring-hydroxylated derivatives (3-hydroxy BP (BP-3OH) and 4-hydroxyBP (BP-4OH)) that have estrogenic activity. BP-4OH was more potent than BP-3OH for promoting estrogen receptor (ER)-mediated transcription and uterotrophic activity, although both of them showed same affinity in ER binding. The reproductive toxicity of BZP was evaluated in a two generation test in which male and female rats, parental (F0) and first generation (F1), were exposed to BZP by feeding diet containing BZP at concentrations of 0 (control), 100, 450 or 2000 ppm. With regard to the effects of BZP on the F0 and F1 parental animals, inhibition of body weight gain and food consumption, significantly elevated renal weights, dilatation of the renal proximal tubules, and regeneration of the proximal tubular epithelium were recognized at doses of 450 ppm and 2000 ppm, along with an increase in hepatic weight and centrilobular hepatocytic hypertrophy, considered as vital adaptive changes, at the doses of 100 ppm or more. Obvious effects on the endocrine system and reproductive toxicological effects were not found even at the dose of 2000 ppm in the F0 or F1 parent. There were no test substance related changes in the estrous cycle, reproductive capability, delivery and lactation, sperm parameters, serum hormone levels, or necropsy findings. As for effects on the offspring, inhibition of body weight gain was observed in both the F1 and F2 males and females of the 2000 ppm group. No effects of BZP treatment were recognized in the number of male and female F1 or F2 pups delivered, viability, anogenital distance (AGD), physical development, the results of reflex and response tests, or on the observation results of external abnormalities. In carcinogenicity studies, BZP had no effect on survival rates or on incidences of neoplasms or nonneoplastic lesions in lifetime studies in mice (110 weeks) and rabbits (160 weeks) of treatment when administered dermally between the flanks of mice or inside the left ear. In regards to genotoxicity, it was not mutagenic in the standard Ames test using various strains of Salmonella typhimurium, the Escherichia coli pol A assay, in the mouse lymphoma cell mutagenicity test; with or without liver S9 metabolic activation enzymes. BZP and its metabolically related benzhydrol and p-benzoylphenol can be bioactivated by P450 2A6 and P450 family 1 enzymes. ECOTOXICITY STUDIES: The lethal and sublethal effects of six monosubstituted derivatives of benzene were measured by using the 7 day test with fathead minnow larvae. The LC50s for larvae were compared to those derived from the acute test. The larvae were more sensitive than juvenile fish, yet the toxicity order of the six monosubstituted derivatives of benzene was the same for both life stages, that is, butylphenylether > benzophenone > toluene = benzene > nitrobenzene > aniline.
Benzophenone is a cholinesterase or acetylcholinesterase (AChE) inhibitor. A cholinesterase inhibitor (or 'anticholinesterase') suppresses the action of acetylcholinesterase. Because of its essential function, chemicals that interfere with the action of acetylcholinesterase are potent neurotoxins, causing excessive salivation and eye-watering in low doses, followed by muscle spasms and ultimately death. Nerve gases and many substances used in insecticides have been shown to act by binding a serine in the active site of acetylcholine esterase, inhibiting the enzyme completely. Acetylcholine esterase breaks down the neurotransmitter acetylcholine, which is released at nerve and muscle junctions, in order to allow the muscle or organ to relax. The result of acetylcholine esterase inhibition is that acetylcholine builds up and continues to act so that any nerve impulses are continually transmitted and muscle contractions do not stop. Among the most common acetylcholinesterase inhibitors are phosphorus-based compounds, which are designed to bind to the active site of the enzyme. The structural requirements are a phosphorus atom bearing two lipophilic groups, a leaving group (such as a halide or thiocyanate), and a terminal oxygen.
来源:Toxin and Toxin Target Database (T3DB)
毒理性
致癌物分类
国际癌症研究机构致癌物:苯甲酮
IARC Carcinogenic Agent:Benzophenone
来源:International Agency for Research on Cancer (IARC)
毒理性
致癌物分类
国际癌症研究机构(IARC)致癌物分类:2B组:可能对人类致癌
IARC Carcinogenic Classes:Group 2B: Possibly carcinogenic to humans
来源:International Agency for Research on Cancer (IARC)
The percutaneous absorption of benzophenone was determined in vivo in monkeys. Absorption through occluded skin was approximately 70% of the applied dose in 24 hr. Under unoccluded conditions skin penetration was reduced to 44%, presumably because of evaporation from the site of application.
/The authors/ gave /benzophenone/ orally ... (100 or 400 mg/kg) ... once per day for 3 days, to ovariectomized Sprague-Dawley (SD) rats, and all rats were killed 24 h after being given the last dose. ... At 24 hr after the last dose, the mean serum concentrations of benzophenone, benzhydrol and p-hydroxybenzophenone in the high-dosed rats were 10.4+/-1.0, 1.5+/-0.3, and 0.7+/-0.2 (mean +/- SE) umol/L, respectively, whereas in the serum of low-dosed rats these compounds were not detected. When a single oral administration of benzophenone (100 or 400 mg/kg) was given to intact female rats, serum concentrations of benzophenone, benzhydrol and p-hydroxybenzophenone increased in a dose-dependent manner 6 hr later. ...
... The percutaneous absorption of the fragrances benzyl acetate and five other benzyl derivatives (benzyl alcohol, benzyl benzoate, benzamide, benzoin and benzophenone) was determined in vivo in monkeys. Absorption through occluded skin was high for all cmpd (approx 70% of the applied dose in 24 hr) and no significant differences between the values for the different cmpd were observed. No correlations were seen between skin penetration of these cmpd and their octanol-water partition coefficients. Under unoccluded conditions skin penetration of the fragrances was reduced and there was great variability between cmpd, presumably because of variations in the rates of evaporation from the site of application.
The present invention relates to oligoesters and their use or the creation of additives. Oligoester containing additives and/or oligoesters themselves may be used for formulating pharmaceutical preparations, cosmetics or personal care products such as shampoos and conditioners. These oligoesters are particularly useful for the creation of multi-purpose additives that can impart conditioning, long substantivity and/or UV protection. Individual oligoesters and oligoester mixtures are described.
Cell adhesion-inhibiting antiinflammatory and immune-suppressive compounds
申请人:Abbott Laboratories
公开号:US20040116518A1
公开(公告)日:2004-06-17
The present invention relates to novel cinnamide compounds that are useful for treating inflammatory and immune diseases and cerebral vasospasm, to pharmaceutical compositions containing these compounds, and to methods of inhibiting inflammation or suppressing immune response in a mammal.
Design, synthesis, and functional assessment of Cmpd-15 derivatives as negative allosteric modulators for the β2-adrenergic receptor
作者:Kaicheng Meng、Paul Shim、Qingtin Wang、Shuai Zhao、Ting Gu、Alem W. Kahsai、Seungkirl Ahn、Xin Chen
DOI:10.1016/j.bmc.2018.03.023
日期:2018.5
The β2-adrenergic receptor (β2AR), a G protein-coupled receptor, is an important therapeutic target. We recently described Cmpd-15, the first small molecule negative allosteric modulator (NAM) for the β2AR. Herein we report in details the design, synthesis and structure-activity relationships (SAR) of seven Cmpd-15 derivatives. Furthermore, we provide in a dose-response paradigm, the details of the
Polystyrene-bound diaryl selenoxide and telluroxide have been prepared, which behaved as mild oxidizingagents for thiols to disulfides, phosphines to phosphine oxides, hydroquinone and catechol to p- and o-benzoquinones, and thioketones to oxo compounds. The telluroxide completed these reactions in shorter periods or under milder conditions than the selenoxide. In addition, they effected novel solvent-dependent
[EN] BICYCLYL-SUBSTITUTED ISOTHIAZOLINE COMPOUNDS<br/>[FR] COMPOSÉS ISOTHIAZOLINE SUBSTITUÉS PAR UN BICYCLYLE
申请人:BASF SE
公开号:WO2014206910A1
公开(公告)日:2014-12-31
The present invention relates to bicyclyl-substituted isothiazoline compounds of formula (I) wherein the variables are as defined in the claims and description. The compounds are useful for combating or controlling invertebrate pests, in particular arthropod pests and nematodes. The invention also relates to a method for controlling invertebrate pests by using these compounds and to plant propagation material and to an agricultural and a veterinary composition comprising said compounds.