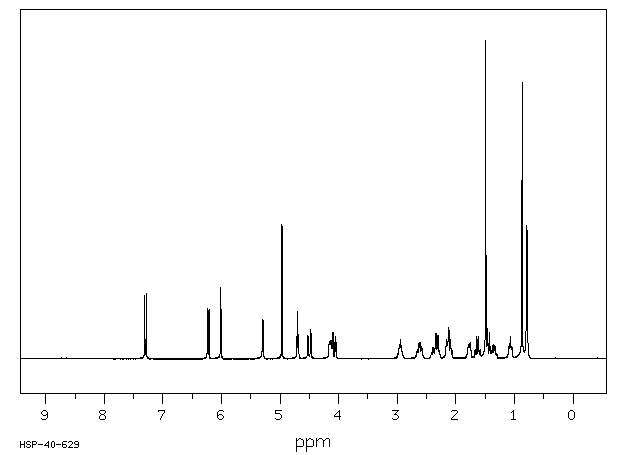
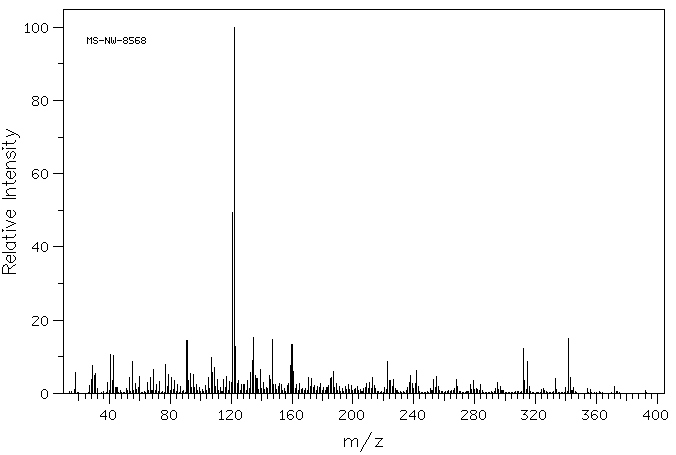
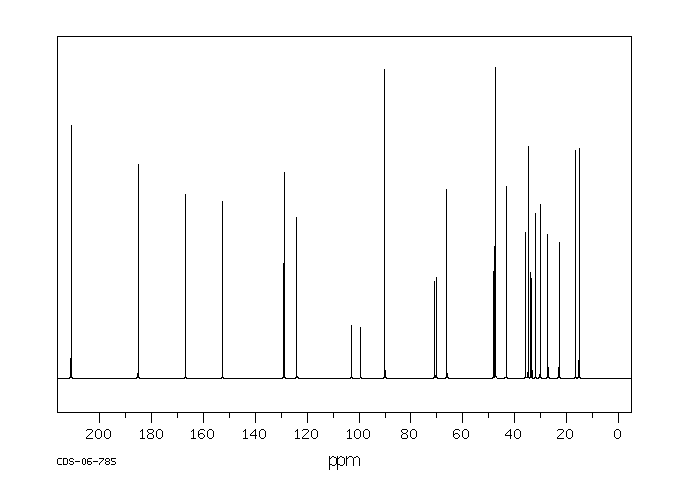
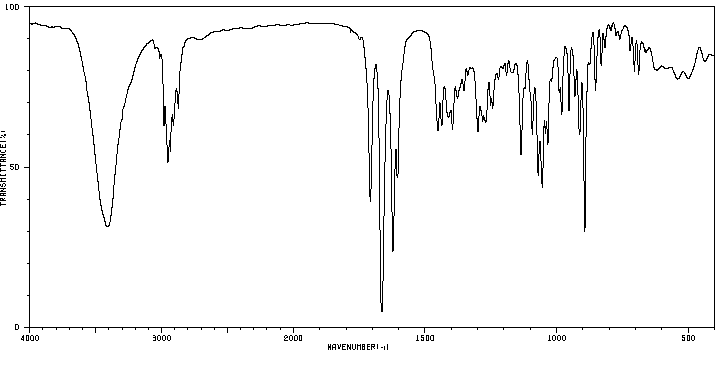
| (8S,9R,10S,11S,13S,14S,16R,17R)-9-氟-11,17-二羟基-10,13,16-三甲基-17-[2-(苯基甲氧基)乙酰基]-6,7,8,11,12,14,15,16-八氢环戊烯并[a]菲-3-酮 |
9αfluoro-11β,17αdihydroxy-21-benzyloxy-16α-methylpregna-1,4-diene-3,20-dione |
150587-07-8 |
C29H35FO5 |
482.592 |
| 去羟米松 |
desoximetasone |
140218-14-0 |
C22H29FO4 |
376.468 |
| 地塞米松17-丙酸酯 |
dexamethasone 17-propionate |
15423-89-9 |
C25H33FO6 |
448.532 |
| —— |
9α-fluoro-11β,17α-dihydroxy-16α-methyl-3-oxoandrosta-1,4-diene-17β-carbothioic acid |
160283-12-5 |
C21H27FO4S |
394.507 |
| 9-氟-11beta,17,21-三羟基-16alpha-甲基孕甾-1,4-二烯-3,20-二酮 17,21-二(丙酸酯) |
betamethasone 17,21 dipropionate |
55541-30-5 |
C28H37FO7 |
504.596 |
| (11beta,16alpha,17alpha)-9-氟-17-(甲酰氧基)-11-羟基-16-甲基-3-氧代雄甾-1,4-二烯-17-羧酸 |
17α-formyloxy-11β-hydroxy-9α-fluoro-16α-methyl-3-oxo-1,4-androstadien-17β-carboxylic acid |
473273-04-0 |
C22H27FO6 |
406.451 |
| 17-氧代地塞米松 |
9α-fluoro-androsta-1,4-diene-11β-hydroxy-16α-methyl-3,17-dione |
1880-61-1 |
C20H25FO3 |
332.415 |
| (8S,9R,10S,11S,13S,14S,16S)-9-氟-11-羟基-10,13,16-三甲基-6,7,8,11,12,14,15,16-八氢环戊烯并[a]菲-3,17-二酮 |
9α-fluoro-11β-hydroxy-16β-methylandrosta-1,4-diene-3,17-dione |
3109-01-1 |
C20H25FO3 |
332.415 |
| 21-氯-9-氟-11beta,17-二羟基-16alpha-甲基孕甾-1,4-二烯-3,20-二酮17-丙酸酯 |
21-chloro-9α-fluoro-11β-hydroxy-16α-methyl-17α-propanoyloxy-1,4-pregnadiene-3,20-dione |
25122-52-5 |
C25H32ClFO5 |
466.977 |
| —— |
tert-butyl (2-(((2-((8S,9R,10S,11S,13S,14S,16R,17R)-9-fluoro-11,17-dihydroxy-10,13,16-trimethyl-3-oxo-6,7,8,9,10,11,12,13,14,15,16,17-dodecahydro-3H-cyclopenta[a]phenanthren-17-yl)-2-oxoethoxy)(hydroxy)phosphoryl)oxy)ethyl)carbamate |
—— |
C29H43FNO10P |
615.633 |
| —— |
11-Dehydrodexamethasone |
2964-81-0 |
C22H27FO5 |
390.452 |
| —— |
9α-fluoro-11β,17α-dihydroxy-16α-methyl-3,20-dioxopregna-1,4-dien-21-yl β-D-galactopyranoside |
92901-23-0 |
C28H39FO10 |
554.61 |
| —— |
(Z)-2-((8S,9R,10S,11S,13S,14S,16R)-9-fluoro-11-hydroxy-10,13,16-trimethyl-3-oxo-3,6,7,8,9,10,11,12,13,14,15,16-dodecahydro-17H-cyclopenta[a]phenanthren-17-ylidene)-2-hydroxyethanaldehyde |
1174035-75-6 |
C22H27FO4 |
374.452 |
| —— |
(8S,9R,10S,11S,13S,14S,16R,17R)-17-(2-(benzo[d]thiazol-2-ylthio)acetyl)-9-fluoro-11,17-dihydroxy-10,13,16-trimethyl-6,7,8,9,10,11,12,13,14,15,16,17-dodecahydro-3H-cyclopenta[a]phenanthren-3-one |
166976-58-5 |
C29H32FNO4S2 |
541.708 |
| 氢化可的松 |
HYDROCORTISONE |
50-23-7 |
C21H30O5 |
362.466 |
| 地塞米松环氧水解物 |
17α,21-dihydroxy-9β,11β-oxido-16α-methylpregna-1,4-diene-3,20-dione |
24916-90-3 |
C22H28O5 |
372.461 |