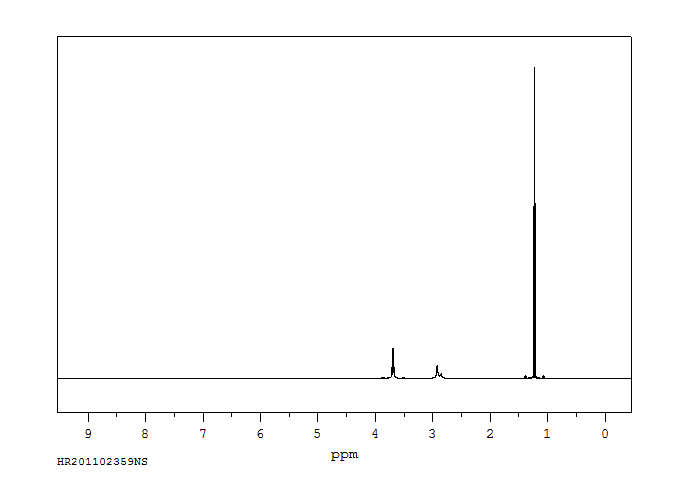
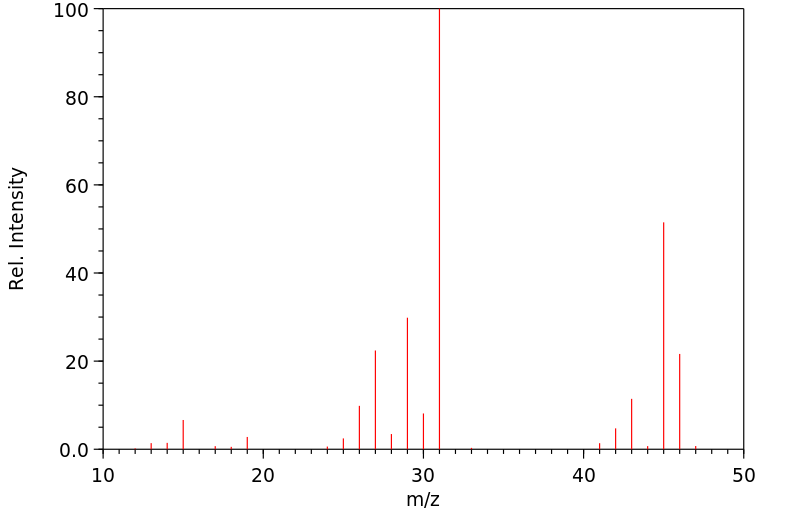
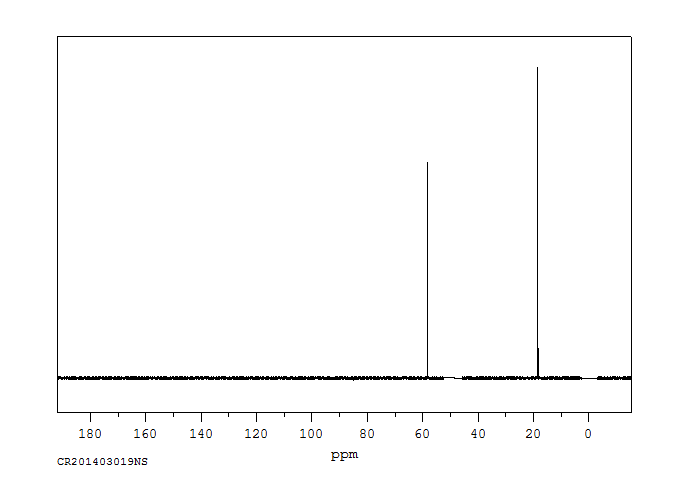
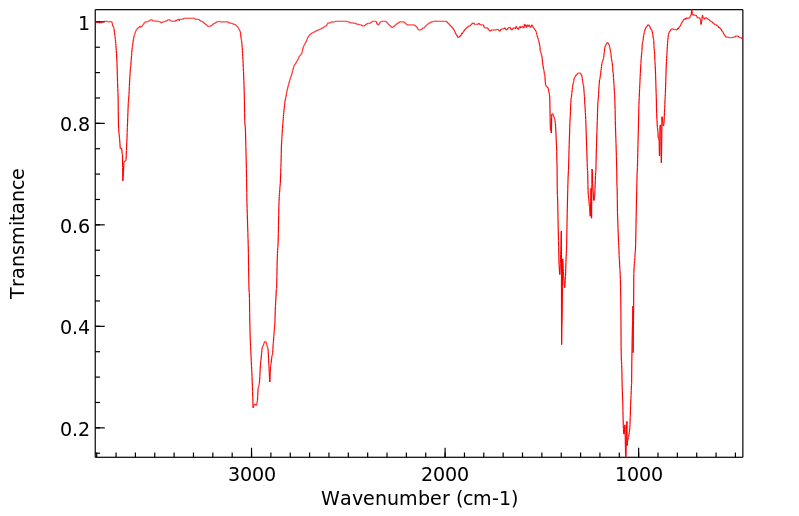
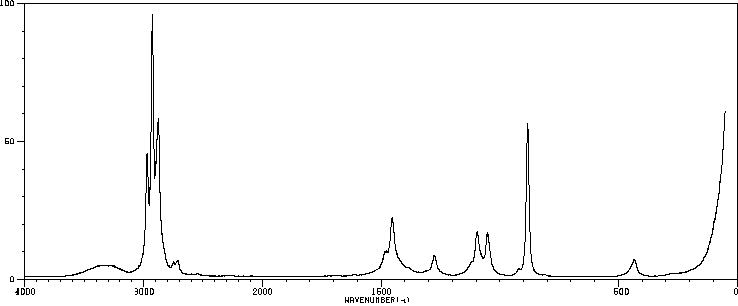
Ethanol appears as a clear colorless liquid with a characteristic vinous odor and pungent taste. Flash point 55°F. Density 6.5 lb / gal. Vapors are heavier than air.
颜色/状态:
Clear, colorless, very mobile liquid
气味:
Pleasant
味道:
Burning
蒸汽密度:
1.59 (NTP, 1992) (Relative to Air)
蒸汽压力:
VP: -73 °C at 1 Pa; -56 °C at 10 Pa; -34 °C at 100 Pa; -7 °C at 1 kPa (all extrapolated); 29.2 °C at 10 kPa; 78.0 °C at 100 kPa
亨利常数:
Henry's Law constant = 5X10-6 atm-cu m/mol at 25 °C
Ethanol metabolism in hepatocytes causes the generation of reactive oxygen species, endoplasmic reticulum stress and alterations in mitochondrial energy and REDOX metabolism. In ethanol-exposed liver disease, autophagy not only acts as a cleanser to remove damaged organelles and cytosolic components, but also selectively clears specific targets such as lipid droplets and damaged mitochondria. Moreover, ethanol appears to play a role in protecting hepatocytes from apoptosis at certain concentrations. This article describes the evidence, function and potential mechanism of autophagy in ethanol-exposed liver disease and the controversy surrounding the effects of ethanol on autophagy.
There have been allegations in the courtroom that elevated serum lactic acid in trauma victims can yield a falsely elevated serum ethanol assay. Most hospitals utilize an indirect method of ethanol measurement where a serum sample is added to a mix of alcohol dehydrogenase and oxidized nicotinamide adenine dinucleotide (NAD+). This allows any ethanol in the patient's serum to be metabolized to acetaldehyde, and in the process results in the reduction of NAD+ to NADH. NADH is then measured using spectrophotometry. The courtroom allegation stems from the concept that oxidation of lactate to pyruvate by lactate dehydrogenase (LDH) results in the same molar-for-molar reduction of NAD+ to NADH, and could therefore theoretically cause patients with elevated lactate and LDH to have a falsely elevated ethanol concentration. Patients with elevated lactic acid and LDH concentrations who presented to a university hospital from 20 April 2015 to 13 December 2015 were identified to provide possible test specimens. If a sufficient amount of serum was available, the sample was used to re-run the lactate and LDH concentration simultaneously with an enzymatic ethanol assay. Any samples that had elevated lactic acid and LDH concentrations on this retesting, and also yielded a positive ethanol concentration, were sent for confirmatory gas chromatography testing of ethanol concentrations. A control group of 20 samples with normal lactate and LDH were included. A total of 37 samples were included in the final analysis. Only 4 patients had an elevated enzymatic ethanol concentration, and all 4 also had a measurable GC ethanol concentration. The lactate in this dataset ranged from 2.4 to 24.2 mmol/L, with a mean of 6.53 mmol/L (normal value 0.5-2.2). The LDH ranged from 242 to 8838 U/L with a mean of 1695 U/L (normal value 122-225 U/L). Twenty control samples were run on patients with normal lactate and LDH, none of which yielded a positive enzymatic ethanol result. This data does not support the contention that an elevated LDH and lactate can yield a false positive serum ethanol result as run by enzymatic ethanol assay in live patients presenting to the emergency department.
Ethanol is metabolized largely by sequential hepatic oxidation, first to acetaldehyde by alcohol dehydrogenase (ADE) and then to acetic acid by aldehyde dehydrogenase (ALDH). Each metabolic step requires NAD+; thus oxidation of 1 mol ethanol (46 g) to 1 mol acetic acid requires 2 mol NAD+ in the liver; indeed, NAD+ availability limits ethanol metabolism to about 8 gr or 10 mL (approximately 170 mmol) per hour in a 70-kg adult, or approximately 120 mg/kg per hour. Thus hepatic ethanol metabolism functionally saturates at relatively low blood levels compared with the high blood ethano levels (BELs) achieved, and ethanol metabolism is a zero-order process (constant amount per unit time). Small amounts of ethanol are excreted in urine, sweat, and breath, but metabolism to acetate accounts to 90-98% of ingested ethanol, mostly owing to hepatic metabolism by ADH and ADLH.
Metabolism of ethanol, propanol, isopropanol, butanol, isobutanol, sec-butanol, and tert-butanol was studied after oral administration in rabbits. Blood pH was on the acid side with propanol, butanol, and isobutanol, and on the alkaline side with isopropanol and sec-butanol, but no change was observed with ethanol and tert-butanol. Butanol and isobutanol had the lowest rate of urinary excretion. Acetaldehyde and acetic acid were detected as the urinary metabolites of ethanol and propanol, whereas isobutyraldehyde and isovaleric acid were the metabolites of isobutanol.
IDENTIFICATION AND USE: Ethanol is a clear, colorless, very mobile liquid. It is used in alcoholic beverages in suitable dilutions, and as a reagent in synthetic organic chemistry and chromatography, as well as industrial and laboratory organic solvent. Other uses are in manufacture of denatured alcohol, pharmaceuticals (rubbing compounds, lotions, tonics, colognes), in perfumery. Octane booster in gasoline. Pharmaceutic aid (solvent). HUMAN STUDIES: Ethanol is a central nervous system (CNS) depressant. It enhances the inhibitory effects of gamma-aminobutyric acid (GABA) at the GABA-A receptor and competitively inhibits the binding of glycine at the N-methyl-d-aspartate receptor (it disrupts excitatory glutaminergic neurotransmission). Ethanol also stimulates release of other inhibitory neurotransmitters, such as dopamine and serotonin. The most common clinical signs of ethanol toxicosis are ataxia, lethargy, vomiting, and recumbency. In more severe cases, hypothermia, disorientation, vocalization, hypotension, tremors, tachycardia, acidosis, diarrhea, respiratory depression, coma, seizures, and death may occur. Alcohol is directly irritating to the stomach and causes vomiting. High ethanol blood levels also stimulate emesis. The concern with vomiting during intoxication is that at high blood ethanol concentrations, the muscles that control the epiglottis become slow to react or even paralyzed. This increases the risk for aspiration. Ethanol intoxication reduces peripheral oxygen delivery and metabolism and causes mitochondrial oxidative dysfunction, potentially resulting in shock or hypoxia in an acutely intoxicated patient. Hypothermia may result from multiple mechanisms. Peripheral vasodilation, CNS depression, ethanol interference with the thermoregulator mechanism, and/or impaired behavioral responses to a cold environment all lead to a lowered body temperature. Moderate ethanol intake appears to reduce the risk of myocardial infarction and other heart diseases. However, high spirits consumption was associated with increased risk of cancer mortality in women. Consumption of alcoholic beverages (beer, in particular) is associated with an increased risk for rectal but not colon cancer. Beer is a commonly consumed alcoholic beverage among reproductive-age adults. Beer drinking males have an increased risk of contributing to pregnancy waste. Women consume beer before and after pregnancy recognition. Binge drinking appears to be a common drinking behavior, and those who binge drink have an increased risk of impaired fetal growth and offspring behavior. Beer consumption by lactating women might temporarily impair motor function of nursing infants. The rate of ethanol metabolism varies among individuals. Studies of twins indicate that interindividual variability in the rate of ethanol metabolism may be genetically controlled. The main pathway for ethanol oxidation in humans is to acetaldehyde via alcohol dehydrogenase pathway. Acetaldehyde is oxidized further to acetic acid by aldehyde dehydrogenase. Asians are known to be sensitive to the health effects of ethanol; the sensitivity has been attributed to different forms of the enzyme acetaldehyde dehydrogenase. Alcohol ingestion by Asians resulted in marked elevations of blood acetaldehyde levels ranging from 0.4 to 3 mg/L, and individuals developed facial flushing and tachycardia as a direct consequence of elevated blood acetaldehyde levels. ANIMAL STUDIES: A drop full-strength ethanol on rabbit eyes causes reversible injury graded only 3 on a scale of 10 after 24 hr. Application of 70% alcohol to rabbit corneas injures and temporarily loosens the corneal epithelium, but the recovery is complete. When rats were dosed with ethanol by oral gavage with 8 to 15 g/kg/day over 4 months and fed a diet containing 25% of total calories as fat, focal necrosis, inflammation, and fibrosis were observed in the liver. Nine baboons fed ethanol at 50% of total calories developed fatty liver, and four animals developed hepatitis within 9 to 12 months. Rabbits exposed to saturated vapors of ethanol for periods ranging from 25 to 365 days developed cirrhosis of the liver. Rats were given a single intraperitoneal dose of diethylnitrosamine followed by treatment with ethanol in drinking water for 12 to 18 months. Ethanol was an effective promoter of liver tumors. Cynomolgus monkeys administered up to 5 g/kg bw ethanol daily on gestation days 20-150 revealed an increase in pregnancy wastage (abortions and still births) but no structural malformation or facial change. Ethanol, and not acetaldehyde, has been implicated as the causative agent of the teratogenic effects in laboratory animals. Oral coadministration of 100 mg/kg of 4-methylpyrazole, an inhibitor of alcohol dehydrogenase, with 6 g/kg of ethanol intraperitoneally on gestation day 10 dramatically increased the embryotoxicity of ethanol in mice. Ethanol is not mutagenic in Salmonella typhimurium strains TA 97, TA 98, TA 100, TA 1535, TA 1537, or TA 1538 in the presence or absence of metabolic activation. In the presence of a metabolic activation system, ethanol is slightly mutagenic to Salmonella strain TA 102, a strain considered to respond to the presence of oxygen radicals. Ethanol did not induce mutations in mouse lymphoma L5178Y TK+/- cells and did not induce micronuclei in Chinese hamster V79 cells in the absence of metabolic activation. No chromosomal aberrations or sister chromatid exchanges were observed in Chinese hamster ovary cells treated with ethanol. ECOTOXICITY STUDIES: The zebrafish were exposed to different concentrations (control, 0.01, 0.1, and 1%) of ethanol from blastula stage to 144 hour-post-fertilization (hpf). No effect on survival was observed except the 1% ethanol group suffered 89% mortality during 108-120 hpf. No developmental defects were observed at the 0.01 and 0.1% concentrations, but significantly higher deformity rates occurred with 1% ethanol. Hyperactivity and less tortuous swimming paths were observed in all ethanol concentrations.
Alcohol binds to the GABA(A) receptors (delta subunit), NMDA receptors, Glycine receptors, Serotonin receptors, Acetylcholine receptors, L-channel calcium channels and GIRK channels. Ethanol acts in the central nervous system primarily by binding to the GABAA receptor, increasing the effects of the inhibitory neurotransmitter GABA. Ethanol within the human body is converted into acetaldehyde by alcohol dehydrogenase. Acetaldehyde is linked to most of the clinical effects of alcohol. It has been shown to increase the risk of developing cirrhosis of the liver and multiple forms of cancer. During the metabolism of alcohol via the respective dehydrogenases, NAD (Nicotinamide adenine dinucleotide) is converted into reduced NAD. Normally, NAD is used to metabolise fats in the liver, and as such alcohol competes with these fats for the use of NAD. Prolonged exposure to alcohol means that fats accumulate in the liver, leading to the term 'fatty liver'. Continued consumption (such as in alcoholism) then leads to cell death in the hepatocytes as the fat stores reduce the function of the cell to the point of death. These cells are then replaced with scar tissue, leading to the condition called cirrhosis.
来源:Toxin and Toxin Target Database (T3DB)
毒理性
致癌性证据
A3;已确认对动物有致癌性,但对人类的相关性未知。
A3; Confirmed animal carcinogen with unknown relevance to humans.
After oral administration, ethanol is absorbed rapidly into the bloodstream from the stomach and small intestines and distributes into total body water (0.5-0.7 L/kg). Peak blood levels occur about 30 minutes after ingestion of ethanol when the stomach is empty. Because absorption occurs more rapidly from the small intestine than from the stomach, delays in gastric emptying (owing, e.g., to the presence of food) slow ethanol absorption. ... After oral consumption of alcohol, first-pass metabolism by gastric and liver alcohol dehydrogenase enzymes leads to lower blood alcohol levels than would be obtained if the same dose were administered intravenously.
The distribution of alcohol between alveolar air and blood depends on its speed of diffusion, and its vapor pressure at the prevailing temp and concentration of alcohol in the lung capillaries. Empirical determinations have yielded rather different values for this distribution ratio, but a commonly accepted value is 1:2100.
Venous blood (orbital sinus) and brain ethanol levels were measured in long sleep and short sleep mice within the first 30 min following ethanol administration (2.5 to 6.0 g/kg). Ethanol was administered ip or intragastrically. For both lines of mice and for every dose, brain ethanol concentrations were significantly greater (as much as 100 mg/dL) than blood ethanol levels for the first 6 min, and peak blood and brain ethanol levels were reached 4 to 6 min after dosing. Approx 6 to 10 min (depending on dose and line of mouse) was required for blood and brain concn to reach equilibrium. At the time of loss of the righting response brain ethanol levels were significantly higher than blood ethanol levels. These results indicate that within the first 6 min after administration of ethanol, blood ethanol level is not suitable for the assessment of brain ethanol content.
The method of Pohorecky and Brick was modified for determination of ethanol concn in rebreathed air of rats. Female Sprague Dawley rats were injected with different doses (1 to 2 g/kg) of ethanol and both arterial blood and rebreathed air samples were collected at various time intervals (15 to 120 min) after administration. A good correlation (r= 0.96) was found between ethanol concn in arterial blood and in rebreathed air; the blood/breath conversion factor was 3241 + or - 55.
Properties and Reactions of Substituted 1,2-Thiazetidine 1,1-Dioxides: Chiral Mono- and Bicyclic 1,2-Thiazetidine 1,1-Dioxides fromα-Amino Acids
作者:Alexandra Meinzer、Andrea Breckel、Bassam Abu Thaher、Nico Manicone、Hans-Hartwig Otto
DOI:10.1002/hlca.200490021
日期:2004.1
New chiral mono- and bicyclic β-sultams, valuable building blocks for drug synthesis, have been prepared from L-Ala, L-Val, L-Leu, L-Ile, L-Phe, L-Cys, L-Ser, L-Thr, and D-penicillamine by transformation of the COOH group into a methylsulfonyl chloride function, followed by cyclization under basic conditions. Selected properties, derivatives, and reactions of the β-sultams are described.
[EN] BENZAMIDE OR BENZAMINE COMPOUNDS USEFUL AS ANTICANCER AGENTS FOR THE TREATMENT OF HUMAN CANCERS<br/>[FR] COMPOSÉS BENZAMIDE OU BENZAMINE À UTILISER EN TANT QU'ANTICANCÉREUX POUR LE TRAITEMENT DE CANCERS HUMAINS
申请人:UNIV TEXAS
公开号:WO2017007634A1
公开(公告)日:2017-01-12
The described invention provides small molecule anti-cancer compounds for treating tumors that respond to cholesterol biosynthesis inhibition. The compounds selectively inhibit the cholesterol biosynthetic pathway in tumor-derived cancer cells, but do not affect normally dividing cells.
The synthesis of 1-chloroalkyl carbonates and their reaction with various type of amines are described. This reaction is useful for the synthesis of carbamate pesticides and for the protection of various amino groups, including amino acids.
The scope of MgI2 as a valuable tool for quantitative and mild chemoselective cleavage of protectinggroups is described here. This novel synthetic approach expands the use of protectinggroups, widens the concept of orthogonality in synthetic processes, and offers a facile opportunity to release compounds from solid supports.