
2-氯-4-甲氧基苯硼酸 | 219735-99-6
中文名称
2-氯-4-甲氧基苯硼酸
中文别名
2-氯-4-茴香醚硼酸;2-氯-4-甲氧基苯基硼酸
英文名称
2-chloro-4-methoxyphenylboronic acid
英文别名
(2-chloro-4-methoxylphenyl)boronic acid;(2-chloro-4-methoxyphenyl)boronic acid

CAS
219735-99-6
化学式
C7H8BClO3
mdl
MFCD03411935
分子量
186.403
InChiKey
LOCGPWGCRVKCFN-UHFFFAOYSA-N
BEILSTEIN
——
EINECS
——
-
物化性质
-
计算性质
-
ADMET
-
安全信息
-
SDS
-
制备方法与用途
-
上下游信息
-
文献信息
-
表征谱图
-
同类化合物
-
相关功能分类
-
相关结构分类
物化性质
-
熔点:190-196°C
-
沸点:343.4±52.0 °C(Predicted)
-
密度:1.32±0.1 g/cm3(Predicted)
-
稳定性/保质期:
避氧化物
计算性质
-
辛醇/水分配系数(LogP):1.91
-
重原子数:12
-
可旋转键数:2
-
环数:1.0
-
sp3杂化的碳原子比例:0.142
-
拓扑面积:49.7
-
氢给体数:2
-
氢受体数:3
安全信息
-
危险等级:IRRITANT
-
危险品标志:Xi
-
安全说明:S26,S36/37/39
-
危险类别码:R36/37/38
-
海关编码:2931900090
-
危险性防范说明:P261,P305+P351+P338
-
危险性描述:H302,H315,H319,H335
SDS
2-氯-4-甲氧基苯硼酸(含有数量不等的酸酐) 修改号码:6
模块 1. 化学品
产品名称: 2-Chloro-4-methoxyphenylboronic Acid (coNTains varying amouNTs of
Anhydride)
修改号码: 6
模块 2. 危险性概述
GHS分类
物理性危害 未分类
健康危害
皮肤腐蚀/刺激 第2级
严重损伤/刺激眼睛 2A类
环境危害 未分类
GHS标签元素
图标或危害标志
信号词 警告
危险描述 造成皮肤刺激
造成严重眼刺激
防范说明
[预防] 处理后要彻底清洗双手。
穿戴防护手套/护目镜/防护面具。
[急救措施] 眼睛接触:用水小心清洗几分钟。如果方便,易操作,摘除隐形眼镜。继续冲洗。
眼睛接触:求医/就诊
皮肤接触:用大量肥皂和水轻轻洗。
若皮肤刺激:求医/就诊。
脱掉被污染的衣物,清洗后方可重新使用。
模块 3. 成分/组成信息
单一物质/混和物 单一物质
化学名(中文名): 2-氯-4-甲氧基苯硼酸(含有数量不等的酸酐)
百分比: ....
CAS编码: 219735-99-6
2-氯-4-甲氧基苯硼酸(含有数量不等的酸酐) 修改号码:6
模块 3. 成分/组成信息
俗名: 2-Chloro-4-methoxybenzeneboronic Acid (coNTains varying amouNTs of
Anhydride)
分子式: C7H8BClO3
模块 4. 急救措施
吸入: 将受害者移到新鲜空气处,保持呼吸通畅,休息。若感不适请求医/就诊。
皮肤接触: 立即去除/脱掉所有被污染的衣物。用大量肥皂和水轻轻洗。
若皮肤刺激或发生皮疹:求医/就诊。
眼睛接触: 用水小心清洗几分钟。如果方便,易操作,摘除隐形眼镜。继续清洗。
如果眼睛刺激:求医/就诊。
食入: 若感不适,求医/就诊。漱口。
紧急救助者的防护: 救援者需要穿戴个人防护用品,比如橡胶手套和气密性护目镜。
模块 5. 消防措施
合适的灭火剂: 干粉,泡沫,雾状水,二氧化碳
特殊危险性: 小心,燃烧或高温下可能分解产生毒烟。
特定方法: 从上风处灭火,根据周围环境选择合适的灭火方法。
非相关人员应该撤离至安全地方。
周围一旦着火:如果安全,移去可移动容器。
消防员的特殊防护用具: 灭火时,一定要穿戴个人防护用品。
模块 6. 泄漏应急处理
个人防护措施,防护用具, 使用个人防护用品。远离溢出物/泄露处并处在上风处。
紧急措施: 泄露区应该用安全带等圈起来,控制非相关人员进入。
环保措施: 防止进入下水道。
控制和清洗的方法和材料: 清扫收集粉尘,封入密闭容器。注意切勿分散。附着物或收集物应该立即根据合适的
法律法规处置。
模块 7. 操作处置与储存
处理
技术措施: 在通风良好处进行处理。穿戴合适的防护用具。防止粉尘扩散。处理后彻底清洗双手
和脸。
注意事项: 如果粉尘或浮质产生,使用局部排气。
操作处置注意事项: 避免接触皮肤、眼睛和衣物。
贮存
储存条件: 保持容器密闭。存放于凉爽、阴暗处。
远离不相容的材料比如氧化剂存放。
包装材料: 依据法律。
模块 8. 接触控制和个体防护
工程控制: 尽可能安装封闭体系或局部排风系统,操作人员切勿直接接触。同时安装淋浴器和洗
眼器。
个人防护用品
呼吸系统防护: 防尘面具。依据当地和政府法规。
手部防护: 防护手套。
眼睛防护: 安全防护镜。如果情况需要,佩戴面具。
皮肤和身体防护: 防护服。如果情况需要,穿戴防护靴。
2-氯-4-甲氧基苯硼酸(含有数量不等的酸酐) 修改号码:6
模块 9. 理化特性
固体
外形(20°C):
外观: 晶体-粉末
颜色: 白色-极淡的黄色
气味: 无资料
pH: 无数据资料
熔点: 202°C
沸点/沸程 无资料
闪点: 无资料
爆炸特性
爆炸下限: 无资料
爆炸上限: 无资料
密度: 无资料
溶解度:
[水] 无资料
[其他溶剂] 无资料
模块 10. 稳定性和反应性
化学稳定性: 一般情况下稳定。
危险反应的可能性: 未报道特殊反应性。
须避免接触的物质 氧化剂
危险的分解产物: 一氧化碳, 二氧化碳, 氧化硼, 氯化氢
模块 11. 毒理学信息
急性毒性: 无资料
对皮肤腐蚀或刺激: 无资料
对眼睛严重损害或刺激: 无资料
生殖细胞变异原性: 无资料
致癌性:
IARC = 无资料
NTP = 无资料
生殖毒性: 无资料
模块 12. 生态学信息
生态毒性:
鱼类: 无资料
甲壳类: 无资料
藻类: 无资料
残留性 / 降解性: 无资料
潜在生物累积 (BCF): 无资料
土壤中移动性
log水分配系数: 无资料
土壤吸收系数 (Koc): 无资料
亨利定律 无资料
constaNT(PaM3/mol):
模块 13. 废弃处置
如果可能,回收处理。请咨询当地管理部门。建议在可燃溶剂中溶解混合,在装有后燃和洗涤装置的化学焚烧炉中
焚烧。废弃处置时请遵守国家、地区和当地的所有法规。
2-氯-4-甲氧基苯硼酸(含有数量不等的酸酐) 修改号码:6
模块 14. 运输信息
联合国分类: 与联合国分类标准不一致
UN编号: 未列明
模块 15. 法规信息
《危险化学品安全管理条例》(2002年1月26日国务院发布,2011年2月16日修订): 针对危险化学品的安全使用、
生产、储存、运输、装卸等方面均作了相应的规定。
模块16 - 其他信息
N/A
模块 1. 化学品
产品名称: 2-Chloro-4-methoxyphenylboronic Acid (coNTains varying amouNTs of
Anhydride)
修改号码: 6
模块 2. 危险性概述
GHS分类
物理性危害 未分类
健康危害
皮肤腐蚀/刺激 第2级
严重损伤/刺激眼睛 2A类
环境危害 未分类
GHS标签元素
图标或危害标志
信号词 警告
危险描述 造成皮肤刺激
造成严重眼刺激
防范说明
[预防] 处理后要彻底清洗双手。
穿戴防护手套/护目镜/防护面具。
[急救措施] 眼睛接触:用水小心清洗几分钟。如果方便,易操作,摘除隐形眼镜。继续冲洗。
眼睛接触:求医/就诊
皮肤接触:用大量肥皂和水轻轻洗。
若皮肤刺激:求医/就诊。
脱掉被污染的衣物,清洗后方可重新使用。
模块 3. 成分/组成信息
单一物质/混和物 单一物质
化学名(中文名): 2-氯-4-甲氧基苯硼酸(含有数量不等的酸酐)
百分比: ....
CAS编码: 219735-99-6
2-氯-4-甲氧基苯硼酸(含有数量不等的酸酐) 修改号码:6
模块 3. 成分/组成信息
俗名: 2-Chloro-4-methoxybenzeneboronic Acid (coNTains varying amouNTs of
Anhydride)
分子式: C7H8BClO3
模块 4. 急救措施
吸入: 将受害者移到新鲜空气处,保持呼吸通畅,休息。若感不适请求医/就诊。
皮肤接触: 立即去除/脱掉所有被污染的衣物。用大量肥皂和水轻轻洗。
若皮肤刺激或发生皮疹:求医/就诊。
眼睛接触: 用水小心清洗几分钟。如果方便,易操作,摘除隐形眼镜。继续清洗。
如果眼睛刺激:求医/就诊。
食入: 若感不适,求医/就诊。漱口。
紧急救助者的防护: 救援者需要穿戴个人防护用品,比如橡胶手套和气密性护目镜。
模块 5. 消防措施
合适的灭火剂: 干粉,泡沫,雾状水,二氧化碳
特殊危险性: 小心,燃烧或高温下可能分解产生毒烟。
特定方法: 从上风处灭火,根据周围环境选择合适的灭火方法。
非相关人员应该撤离至安全地方。
周围一旦着火:如果安全,移去可移动容器。
消防员的特殊防护用具: 灭火时,一定要穿戴个人防护用品。
模块 6. 泄漏应急处理
个人防护措施,防护用具, 使用个人防护用品。远离溢出物/泄露处并处在上风处。
紧急措施: 泄露区应该用安全带等圈起来,控制非相关人员进入。
环保措施: 防止进入下水道。
控制和清洗的方法和材料: 清扫收集粉尘,封入密闭容器。注意切勿分散。附着物或收集物应该立即根据合适的
法律法规处置。
模块 7. 操作处置与储存
处理
技术措施: 在通风良好处进行处理。穿戴合适的防护用具。防止粉尘扩散。处理后彻底清洗双手
和脸。
注意事项: 如果粉尘或浮质产生,使用局部排气。
操作处置注意事项: 避免接触皮肤、眼睛和衣物。
贮存
储存条件: 保持容器密闭。存放于凉爽、阴暗处。
远离不相容的材料比如氧化剂存放。
包装材料: 依据法律。
模块 8. 接触控制和个体防护
工程控制: 尽可能安装封闭体系或局部排风系统,操作人员切勿直接接触。同时安装淋浴器和洗
眼器。
个人防护用品
呼吸系统防护: 防尘面具。依据当地和政府法规。
手部防护: 防护手套。
眼睛防护: 安全防护镜。如果情况需要,佩戴面具。
皮肤和身体防护: 防护服。如果情况需要,穿戴防护靴。
2-氯-4-甲氧基苯硼酸(含有数量不等的酸酐) 修改号码:6
模块 9. 理化特性
固体
外形(20°C):
外观: 晶体-粉末
颜色: 白色-极淡的黄色
气味: 无资料
pH: 无数据资料
熔点: 202°C
沸点/沸程 无资料
闪点: 无资料
爆炸特性
爆炸下限: 无资料
爆炸上限: 无资料
密度: 无资料
溶解度:
[水] 无资料
[其他溶剂] 无资料
模块 10. 稳定性和反应性
化学稳定性: 一般情况下稳定。
危险反应的可能性: 未报道特殊反应性。
须避免接触的物质 氧化剂
危险的分解产物: 一氧化碳, 二氧化碳, 氧化硼, 氯化氢
模块 11. 毒理学信息
急性毒性: 无资料
对皮肤腐蚀或刺激: 无资料
对眼睛严重损害或刺激: 无资料
生殖细胞变异原性: 无资料
致癌性:
IARC = 无资料
NTP = 无资料
生殖毒性: 无资料
模块 12. 生态学信息
生态毒性:
鱼类: 无资料
甲壳类: 无资料
藻类: 无资料
残留性 / 降解性: 无资料
潜在生物累积 (BCF): 无资料
土壤中移动性
log水分配系数: 无资料
土壤吸收系数 (Koc): 无资料
亨利定律 无资料
constaNT(PaM3/mol):
模块 13. 废弃处置
如果可能,回收处理。请咨询当地管理部门。建议在可燃溶剂中溶解混合,在装有后燃和洗涤装置的化学焚烧炉中
焚烧。废弃处置时请遵守国家、地区和当地的所有法规。
2-氯-4-甲氧基苯硼酸(含有数量不等的酸酐) 修改号码:6
模块 14. 运输信息
联合国分类: 与联合国分类标准不一致
UN编号: 未列明
模块 15. 法规信息
《危险化学品安全管理条例》(2002年1月26日国务院发布,2011年2月16日修订): 针对危险化学品的安全使用、
生产、储存、运输、装卸等方面均作了相应的规定。
模块16 - 其他信息
N/A
制备方法与用途
反应信息
-
作为反应物:描述:2-氯-4-甲氧基苯硼酸 在 N-甲基吗啉 、 二(三叔丁基膦)钯 、 2-氯-4,6-二甲氧基-1,3,5-三嗪 、 水 、 cesium fluoride 、 lithium hydroxide 作用下, 以 四氢呋喃 、 1,4-二氧六环 、 甲醇 、 水 、 N,N-二甲基甲酰胺 、 甲苯 为溶剂, 反应 25.0h, 生成 3-(2-chloro-4-methoxyphenyl)-4-cyano-5-morpholin-4-yl-thiophene-2-carboxylic acid amide参考文献:名称:Development of Scalable Syntheses of Selective PI3K inhibitors摘要:On the basis of a more practical and scalable route to an iodothiophene, an efficient and reliable synthesis has been developed for three selective PI3K inhibitors. From this advanced intermediate, the three title compounds were each prepared in five additional steps. Key learnings also include: high throughput experimentation (HTE) screening toward a more robust Suzuki coupling, a more efficient triazole synthesis, and an acid/base cleanup developed to purify the final compounds. The final enabled synthesis required no column chromatography.DOI:10.1021/op100286g
-
作为产物:参考文献:名称:Imidazopyridines for the treatment of neurological disorders摘要:促肾上腺皮质激素释放因子(CRF)拮抗剂的化学式(I)及其在治疗精神障碍和神经系统疾病、焦虑相关疾病、创伤后应激障碍、上核性麻痹和进食障碍以及治疗与哺乳动物中的心理障碍和应激相关的结肠过敏性疾病、免疫、心血管或与心脏相关疾病有关的用途。公开号:US06362180B1
文献信息
-
Pyrimidine Derivatives As HSP90 Inhibitors申请人:Chessari Gianni公开号:US20090215777A1公开(公告)日:2009-08-27The invention provides a compound for use as an inhibitor of Hsp90, the compound having the formula (I): or salts, tautomers, solvates or N-oxides thereof; wherein: A is N or a group CR 3 ; R 1 is a monocyclic or bicyclic carbocyclic or heterocyclic ring of 5 to 10 ring members of which up to two ring members may be heteroatoms selected from N, O and S and the remainder are carbon atoms, the carbocyclic or heterocyclic ring being optionally substituted by one or more substituent groups independently selected from R 10 ; and R 2 , R 3 and R 10 are as defined in the claims.该发明提供了一种化合物,用作Hsp90的抑制剂,该化合物具有以下式(I):或其盐、互变异构体、溶剂合物或N-氧化物;其中:A为N或CR基团;R1为由5至10个环成员组成的单环或双环碳环或杂环,其中最多两个环成员可以是从N、O和S中选择的杂原子,其余为碳原子,碳环或杂环可以选择地被一个或多个取代基独立地选择自R10的取代基取代;而R2、R3和R10如权利要求中所定义。
-
ANTI-VIRAL COMPOUNDS, COMPOSITIONS, AND METHODS OF USE申请人:Leivers Martin Robert公开号:US20090226398A1公开(公告)日:2009-09-10Disclosed are compounds and compositions of Formula (I), pharmaceutically acceptable salts and solvates thereof, and their preparation and uses for treating viral infections mediated at least in part by a virus in the Flaviviridae family of viruses.披露了公式(I)的化合物和组合物、药物可接受的盐和溶剂化物,以及它们的制备和使用,用于治疗至少部分由黄病毒科病毒家族中的病毒介导的病毒感染。
-
[EN] PROTEIN KINASE INHIBITORS FOR PROMOTING LIVER REGENERATION OR REDUCING OR PREVENTING HEPATOCYTE DEATH<br/>[FR] INHIBITEURS DE PROTÉINE KINASE PERMETTANT DE FAVORISER LA RÉGÉNÉRATION DU FOIE, OU DE RÉDUIRE OU DE PRÉVENIR LA MORT DES HÉPATOCYTES申请人:HEPAREGENIX GMBH公开号:WO2020016243A1公开(公告)日:2020-01-23The invention relates to compounds of formula (I) which are inhibitors of MKK4 (mitogen-activated protein kinase kinase 4) and their use in promoting liver regeneration or reducing or preventing hepatocyte death. The compounds selectively inhibit protein kinase MKK4 over protein kinases JNK and MKK7. (I), wherein R x, R y , R z and R zz are selected from: a) R x and R y are F and R z and R zz are H; b) R x, R y and R zz are independently halogen and R z is H; c) R x R z and R zz are independently halogen and R y is H; and d) R x R y and R z are independently halogen and R zz is H
-
Niacin receptor agonists, compositions containing such compounds and methods of treatment申请人:Raghavan Subharekha公开号:US20060293364A1公开(公告)日:2006-12-28The present invention encompasses compounds of Formula I: as well as pharmaceutically acceptable salts and hydrates thereof, that are useful for treating atherosclerosis, dyslipidemias and the like. Pharmaceutical compositions and methods of use are also included.本发明涵盖了Formula I的化合物: 以及其药用可接受的盐和水合物,用于治疗动脉粥样硬化、血脂异常等疾病。药物组合物和使用方法也包括在内。
-
Design and synthesis of 1H-pyrazolo[3,4-b]pyridines targeting mitogen-activated protein kinase kinase 4 (MKK4) - A promising target for liver regeneration作者:Bent Pfaffenrot、Philip Klövekorn、Michael Juchum、Roland Selig、Wolfgang Albrecht、Lars Zender、Stefan A. LauferDOI:10.1016/j.ejmech.2021.113371日期:2021.6vemurafenib (8), that showed a high off-target affinity to MKK4 in an initial screening, we followed a scaffold-hopping approach, changing the core heterocycle from 1H-pyrrolo[2,3-b]pyridine to 1H-pyrazolo[2,3-b]pyridine (10). Affinity to MKK4 could be conserved while the selectivity against off-target protein kinases was slightly improved. Further modifications led to 58 and 59 showing high affinity目前,治疗肝功能衰竭的治疗选择非常有限。由于最近通过体内RNAi 实验发现丝裂原活化蛋白激酶激酶 4 (MKK4)是肝细胞再生的主要调节因子,因此我们致力于开发一种针对该蛋白激酶的小分子。从已获批的 BRAF V600E抑制剂 vemuRAfenib ( 8 )开始,该抑制剂在初始筛选中显示出对 MKK4 的高脱靶亲和力,我们采用了支架跳跃方法,将核心杂环从 1 H -吡咯 [2,3- b ]吡啶到 1 H-吡唑并[2,3- b ]吡啶(10)。对 MKK4 的亲和力可以得到保留,而对脱靶蛋白激酶的选择性略有提高。进一步的修改导致58和59在低纳摩尔范围内显示出对 MKK4 的高亲和力,以及来自必需抗靶点(MKK7、JNK1)和脱靶点(BRAF、MAP4K5、ZAK)的强制性多参数优化的优异选择性。 MKK4 通路。在此我们报告了此类中的第一个选择性 MKK4 抑制剂。
表征谱图
-
氢谱1HNMR
-
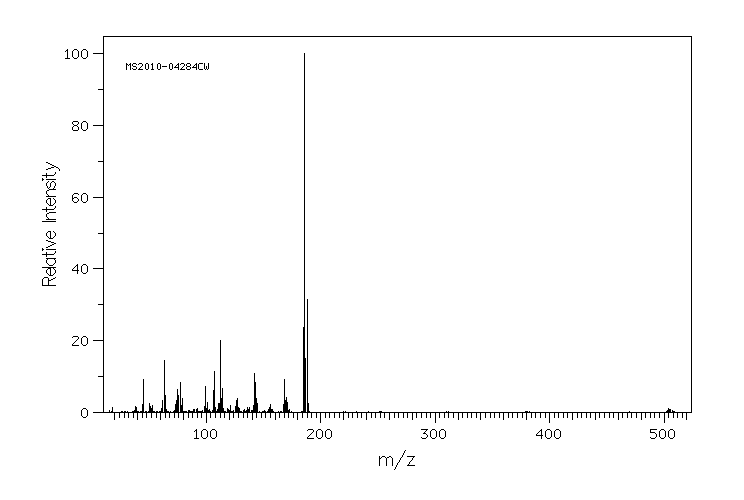
质谱MS
-
碳谱13CNMR
-
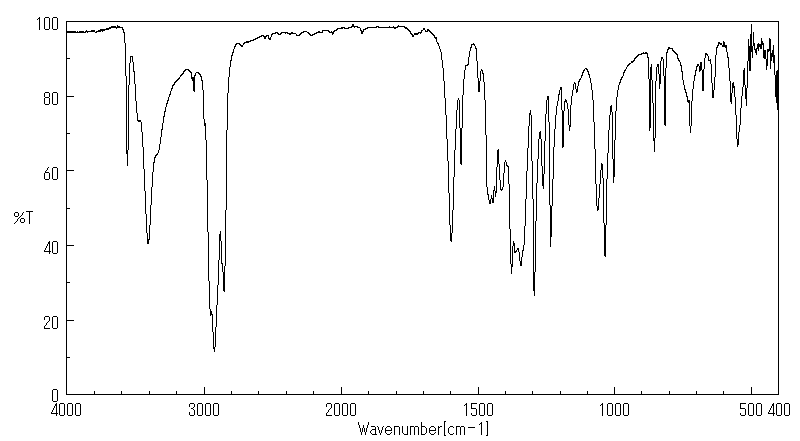
红外IR
-
拉曼Raman
-
峰位数据
-
峰位匹配
-
表征信息
同类化合物
(R)-3-(叔丁基)-4-(2,6-二异丙氧基苯基)-2,3-二氢苯并[d][1,3]氧杂磷杂环戊烯
(2S,3R)-3-(叔丁基)-2-(二叔丁基膦基)-4-甲氧基-2,3-二氢苯并[d][1,3]氧杂磷杂戊环
(2S,2''S,3S,3''S)-3,3''-二叔丁基-4,4''-二甲氧基-2,2'',3,3''-四氢-2,2''-联苯并[d][1,3]氧杂磷杂戊环
(2R,2''R,3R,3''R)-3,3''-二叔丁基-4,4''-二甲氧基-2,2'',3,3''-四氢-2,2''-联苯并[d][1,3]氧杂磷杂戊环
(2-氟-3-异丙氧基苯基)三氟硼酸钾
(+)-6,6'-{[(1R,3R)-1,3-二甲基-1,3基]双(氧)}双[4,8-双(叔丁基)-2,10-二甲氧基-丙二醇
麦角甾烷-6-酮,2,3,22,23-四羟基-,(2a,3a,5a,22S,23S)-
鲁前列醇
顺式6-(对甲氧基苯基)-5-己烯酸
顺式-铂戊脒碘化物
顺式-四氢-2-苯氧基-N,N,N-三甲基-2H-吡喃-3-铵碘化物
顺式-4-甲氧基苯基1-丙烯基醚
顺式-2,4,5-三甲氧基-1-丙烯基苯
顺式-1,3-二甲基-4-苯基-2-氮杂环丁酮
非那西丁杂质7
非那西丁杂质3
非那西丁杂质22
非那西丁杂质18
非那卡因
非布司他杂质37
非布司他杂质30
非布丙醇
雷诺嗪
阿达洛尔
阿达洛尔
阿莫噁酮
阿莫兰特
阿维西利
阿索卡诺
阿米维林
阿立酮
阿曲汀中间体3
阿普洛尔
阿普斯特杂质67
阿普斯特中间体
阿普斯特中间体
阿托西汀EP杂质A
阿托莫西汀杂质24
阿托莫西汀杂质10
阿托莫西汀EP杂质C
阿尼扎芬
阿利克仑中间体3
间苯胺氢氟乙酰氯
间苯二酚二缩水甘油醚
间苯二酚二异丙醇醚
间苯二酚二(2-羟乙基)醚
间苄氧基苯乙醇
间甲苯氧基乙酸肼
间甲苯氧基乙腈
间甲苯异氰酸酯
相关功能分类

联系我们



关注我们

公众号
