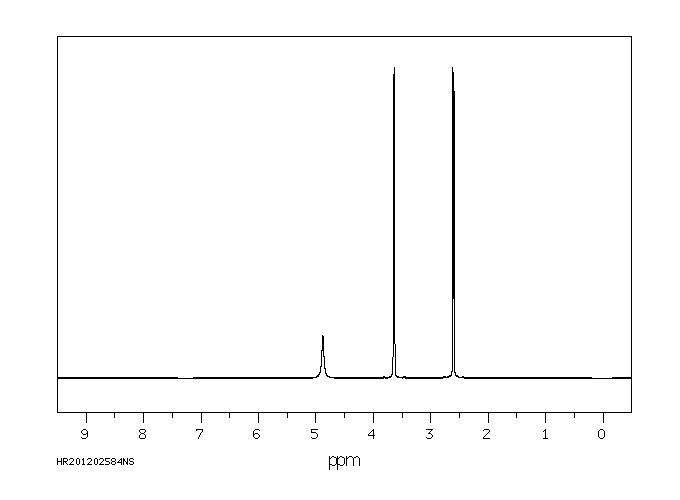
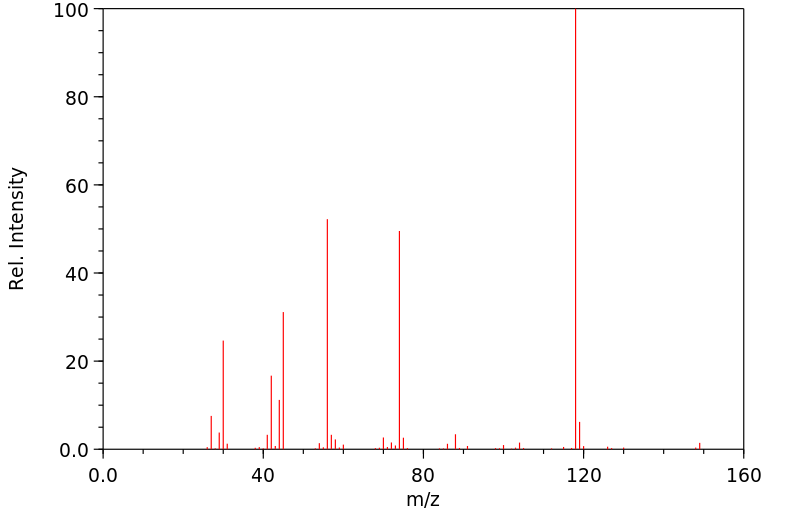
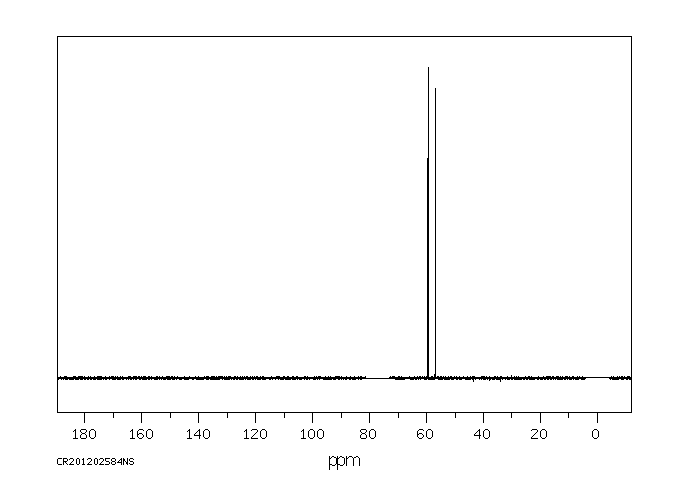
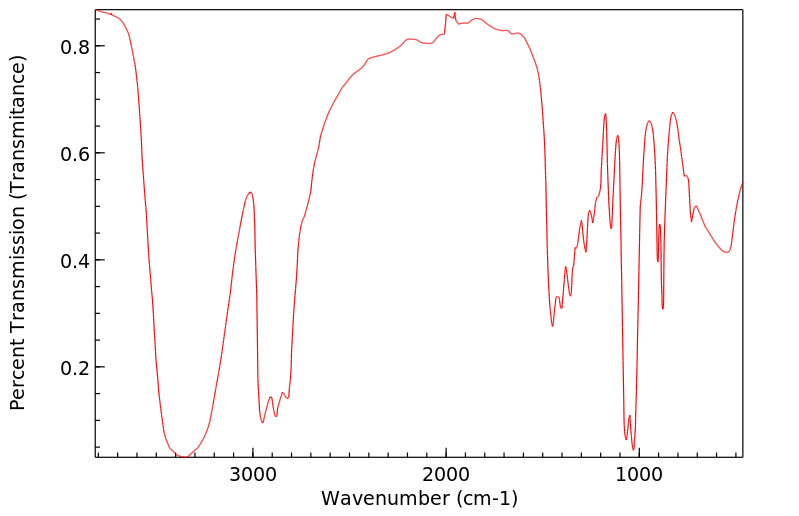
Trolamine is excreted mostly as the unchanged compound. No diethanolamine or ethanolamine has been found. Very small amounts of trolamine glucuronide have been detected but not quantified.
After multiple oral administration to male and female rats, triethanolamine was mainly excreted unchanged. The urinary and fecal excretion ratio of unchanged triethanolamine remained constant throughout the treatment period (for five to six days) in both males and females. A small amount of triethanolamine (1.4-2.7%) was excreted as glucuronide conjugates.
The biotransformation of 14(C)triethanolamine to monoethanolamine and diethanolamine was specifically investigated in mice after both intravenous and dermal treatments. Neither of the hypothetical metabolites was detected in urine (by mass spectral analysis), whereas more than 95% of the radioactivity detected in urine was identified as unchanged triethanolamine. In vitro, triethanolamine had an inhibitory effect on the incorporation of 32(P)phosphate into phospholipids from rabbit and human tissues. Cytochrome P450 monooxygenasedependent oxidative N-dealkylation of triethanolamine does occur in microorganisms, with formation of diethanolamine, ethanolamine and glyoxylate as reaction products.
To determine potential nitrosation of triethanolamine (TEA) to N-nitrosodiethanolamine (NDELA) at different physiological conditions of the GI tract, in vitro NDELA formation was examined in aqueous reaction mixtures at several pHs (2-10) adjusted with acetic, sulfuric or hydrochloric acids or in cultures of mouse cecal microflora incubated. In vivo NDELA formation was also determined in blood, ingesta, and urine of female B6C3F1 mice after repeated dermal, most relevant human route, or single oral exposure to 1000 mg/kg TEA in the presence of high oral dosages of NaNO(2). Appropriate diethanolamine (DEA) controls were included to account for this impurity in the TEA used. Samples were analyzed for NDELA using GC/MS. The highest degree of nitrosation of TEA to NDELA (approximately 3%) was observed in the in vitro cultures at pH 4 and acetic acid with lower amounts obtained using sulphuric acid ( pproximately 1.3%) and hydrochloric acid (approximately 1.2%). At pH 7, <1% of the TEA was nitrosated to NDELA and at pH 2 (HCl) or pH 10 (NaOH) no NDELA was found above the limit of detection. In incubated cultures containing cecal microflora and nutrient broth, only 0.68% of TEA was nitrosated to NDELA. No NDELA was formed in rats repeatedly dermally dosed with TEA at the limits of detection in blood (0.001 ug/mL, ppm), ingesta (0.006 ug/mL, ppm), and urine (0.47 ug/mL, ppm). Levels of NDELA measured in blood and ingesta after a single oral dose of TEA and NaNO(2) were less than those in DEA controls. These findings in toto confirm the lack of any significant formation of NDELA from TEA in vivo.
IDENTIFICATION AND USE: Triethanolamine (TEA) is a colorless, or pale yellow viscous liquid. Triethanolamine is used in the manufacture of emulsifiers and dispersing agents for textile specialties, agricultural chemicals, waxes, mineral and vegetable oils, paraffin, polishes, cutting oils, petroleum demulsifiers, and cement additives. It is an intermediate for resins, plasticizers, and rubber chemicals. It is used as a lubricant in the textile industry, as a humectant and softening agent for hides, as an alkalizing agent and surfactant in pharmaceuticals, as an absorbent for acid gases, and in organic syntheses. It can be useful for rapid detection and identification of chemical warfare agents. TEA has been tested as experimental therapy. HUMAN EXPOSURE AND TOXICITY: Triethanolamine produce mild skin irritation only in concentrations above 5%. It has not been shown to be a sensitizer. Triethanolamine has been identified as causing allergic contact dermatitis, erythematous vesicular lesions, eczema, contact dermatitis, and irritation in workers exposed to triethanolamine in their occupations. A total of 1,357 patients suspected of having allergic eczematous contact dermatitis were patch-tested with triethanolamine. Positive tests were obtained in 41 of these 1,357 patients. The ingestion of several ounces of triethanolamine can probably be tolerated by man, but unless the liquid is partly neutralized with acid, alkali burns of the mouth, pharynx and esophagus are likely. Three cases of occupational asthma caused by ethanolamines were summarized. The three cases share one common feature: exposure to triethanolamines occurred at temperatures higher than that of the ambient air. This agrees with the view that significant inhalation exposure to ethanolamines does not occur when the compounds are used under ambient conditions. ANIMAL STUDIES: Applications of 5 or 10% solution to rabbit or rat skin did not produce irritation. TEA was tested by application of a drop to rabbit eyes. It caused moderate, presumably transient injury, graded 5 on a scale of 1 to 10 after 24 hr, and in another test caused negligible irritation. In a 90-day subacute feeding study with rats, the max dose producing no effect was 80 mg/kg. Microscopic lesions and deaths occurred at 730 mg/kg, and 107 mg/kg produced alterations in liver and kidney weights. Fourteen day repeated dose studies of TEA in rats and mice were performed by inhalation, drinking water, or dermal routes of exposure. Exposures for both species in the inhalation study were 0, 125, 250, 50, 1000 or 2000 mg/cu m, 6 hr/day, 5 days/wk, for 2 wk (10 exposures). The only histopathologic observation was a minimal acute inflammation of the laryngeal submucosa in rats and mice. In the oral study, concentrations of triethanolamine in drinking water (adjusted to pH 7.4) were 0, 500, 1000, 2000, 4000, and 8000 mg/100 mL. Water consumption was significantly reduced in the 4 and 8% dose groups of rats and mice. No compound-related gross or microscopic lesions were observed in the liver or kidneys of rats; cytoplasmic vacuolization of hepatocytes was observed in the high dose groups of male and female mice. Dose levels of triethanolamine in the dermal study were 0, 140, 280, 560, 1130 and 2250 mg/kg for rats and 0, 210, 430, 840, 1690, and 3370 mg/kg for mice. Triethanolamine was applied as the undiluted compound, 5 days/wk for 2 wk. Chronic active necrotizing inflammation of the skin at the application site was observed at a greater frequency and severity in dosed rats than in dosed mice. A Chernoff-Kavlock teratogenicity screening test was performed using mated female mice, in which the animals were dosed by gavage with 1125 mg/kg/day triethanolamine on days 6-15 of gestation. No adverse developmental effects were observed. In a battery of short-term tests, triethanolamine did not induce mutations in bacteria (Salmonella typhimurium strains TA98, TA100, TA1535, TA1537, and TA1538 or Escherichia coli strains WP2 and WP2 uvrA in the presence or absence of S-9 fractions prepared from livers of Aroclor-induced rats), mitotic gene conversion in Saccharomyces cerevisae JD1 cells, or chromosomal damage in cultured rat liver RAL4 cells. Triethanolamine was inactive in inducing revertants to histidine prototrophy in the excision repair deficient Bacillus subtilis strain TKJ5211 with or without rat liver S-9 preparations. In 2-year NTP dermal study, there was equivocal evidence of carcinogenic activity of triethanolamine in male mice based on the occurrence of liver hemangiosarcoma. There was some evidence of carcinogenic activity in female mice based on increased incidences of hepatocellular adenoma. Exposure to triethanolamine by dermal application resulted in increased incidences of eosinophilic focus of the liver in males and females. Dosed mice developed treatment-related nonneoplastic lesions at the site of application. ECOTOXICITY STUDIES: TEA may produce potential acute, sub-chronic and chronic toxicity effects in aquatic species.
Evaluation: There is inadequate evidence in humans for the carcinogenicity of triethanolamine. There is inadequate evidence in experimental animals for the carcinogenicity of triethanolamine. Overall evaluation: Triethanolamine is not classifiable as to its carcinogenicity to humans (Group 3).
Dermal absorption of trolamine increases with the dose. This has been found to range from 19-28% in rats with doses of 68-276 mg/kg in 190 μL of acetone without occlusion and from 60-80% in mice with doses of 79-1120 mg/kg in the same volume of acetone.
When orally administered to rats, the 53% of the trolamine dose was found to be excreted in the urine and 20% in the feces. 98% was excreted in the urine with intravenous administration.
The elimination of 14(C)triethanolamine from the blood of mice administered 1.0 mg/kg bw iv showed first-order biphasic kinetics with a rapid (0.58-hr half life) and a slow phase (10.2-hr half-life). The slow phase half-lives for elimination of triethanolamine in mice after dermal exposure to 1000 and 2000 mg/kg bw in acetone were 9.7 hr and 18.6 hr. Skin absorption rates (as blood concentration-time curves) after dermal application of aqueous and neat 14(C)triethanolamine to mouse skin (2000 mg/kg bw, enclosed by a glass ring) showed no significant change with the use of water as the vehicle.
In a dermal pharmacokinetic study, (14)C-triethanolamine was absorbed more slowly and less extensively in F344 rats than in C3H/HeJ mice. 48 hr after dermal application of (14)C-triethanolamine to mice (1,000 mg/kg dose), about 60% of the radioactivity was recovered from the urine and about 20% was recovered in the feces; less than 10% of the radioactivity was found in skin at the site of application. It was concluded that triethanolamine does not undergo extensive biotransformation in mice, since greater than 95% of the radioactivity recovered from the urine was identified as the parent compound.
Triethanolamine was rapidly absorbed in orally dosed rats, and subsequently excreted mainly as unchanged parent compound in the urine. 24 hr after oral administration of triethanolamine (single dose of 2-3 mg/kg), 53% and 20% of the administered dose was recovered as the parent compound in the urine and feces, respectively.
1.周国泰,化学危险品安全技术全书,化学工业出版社,1997 2.国家环保局有毒化学品管理办公室、北京化工研究院合编,化学品毒性法规环境数据手册,中国环境科学出版社.1992 3.Canadian Centre for Occupational Health and Safety,CHEMINFO Database.1998 4.Canadian Centre for Occupational Health and Safety, RTECS Database, 1989
The invention relates to novel 3-amino pyrrolidine derivatives, as well as methods for modulating calcium channel activity and for treating conditions associated with calcium channel function. In particular, the compounds generally contain at least one benzhydril moiety, and are useful in treating conditions which benefit from blocking calcium ion channels.
[EN] AZA PYRIDONE ANALOGS USEFUL AS MELANIN CONCENTRATING HORMONE RECEPTOR-1 ANTAGONISTS<br/>[FR] ANALOGUES D'AZAPYRIDONE UTILES COMME ANTAGONISTES DU RÉCEPTEUR 1 DE L'HORMONE CONCENTRANT LA MÉLANINE
申请人:BRISTOL MYERS SQUIBB CO
公开号:WO2010104818A1
公开(公告)日:2010-09-16
MCHR1 antagonists are provided having the following Formula (I): A1 and A2 are independently C or N; E is C or N; Q1, Q2, and Q3 are independently C or N provided that at least one of Q1, Q2, and Q3 is N but not more than one of Q1, Q2, and Q3 is N; D1 is a bond, -CR8R9 X-, -XCR8R9-, -CHR8CHR9-, -CR10=CR10'-, -C≡C-, or 1,2-cyclopropyl; X is O, S or NR11; R1, R2, and R3 are independently selected from the group consisting of hydrogen, halogen, lower alkyl, lower cycloalkyl, -CF3, -OCF3, -OR12 and -SR12; G is O, S or -NR15; D2 is lower alkyl, lower cycloalkyl, lower alkylcycloalkyl, lower cycloalkylalkyl, lower cycloalkoxyalkyl or lower alkylcycloalkoxy or when G is NR15, G and D2 together may optionally form an azetidine, pyrrolidine or piperidine ring; Z1 and Z2 are independently hydrogen, lower alkyl, lower cycloalkyl, lower alkoxy, lower cycloalkoxy, halo, -CF3, -OCONR14R14', -CN, -CONR14R14', -SOR12, -SO2R12, -NR14COR14', -NR14CO2R14', -CO2R12, NR14SO2R12 or COR12; R5, R6, and R7 are independently selected from the group consisting of hydrogen lower alkyl, lower cycloalkyl, -CF3, -SR12, lower alkoxy, lower cycloalkoxy, -CN, -CONR14R14', SOR12, SO2R12, NR14COR14', NR14CO2R12, CO2R12, NR14SO2R12 and -COR12; R8, R9, R10, R10', R11 are independently hydrogen or lower alkyl; R12 is lower alkyl or lower cycloalkyl; R14 and R14' are independently H, lower alkyl, lower cycloalkyl or R14 and R14' together with the N to which they are attached form a ring having 4 to 7 atoms; and R15 is independently selected from the group consisting of hydrogen and lower alkyl. Such compounds are useful for the treatment of MCHR1 mediated diseases, such as obesity, diabetes, IBD, depression, and anxiety.
[EN] SULFINYLPYRIDINES AND THEIR USE IN THE TREATMENT OF CANCER<br/>[FR] SULFINYLPYRIDINES ET LEUR UTILISATION DANS LE TRAITEMENT DU CANCER
申请人:OBLIQUE THERAPEUTICS AB
公开号:WO2018146468A1
公开(公告)日:2018-08-16
There is provided compounds of formula I (I) or pharmaceutically-acceptable salts thereof, wherein L, R1, R2, R3, R4 and n have meanings provided in the description, which compounds are useful in the treatment of cancers.
The present invention relates to oligoesters and their use or the creation of additives. Oligoester containing additives and/or oligoesters themselves may be used for formulating pharmaceutical preparations, cosmetics or personal care products such as shampoos and conditioners. These oligoesters are particularly useful for the creation of multi-purpose additives that can impart conditioning, long substantivity and/or UV protection. Individual oligoesters and oligoester mixtures are described.
PYRAZOLO[1,5a]PYRIMIDINE DERIVATIVES AS IRAK4 MODULATORS
申请人:Arora Nidhi
公开号:US20120015962A1
公开(公告)日:2012-01-19
Compounds of the formula I or II:
wherein X, m, Ar, R
1
and R
2
are as defined herein. The subject compounds are useful for treatment of IRAK-mediated conditions.