
2,3-二甲氧基-5-磺酰胺基苯甲酸 | 66644-80-2
中文名称
2,3-二甲氧基-5-磺酰胺基苯甲酸
中文别名
5-(氨基磺酰)-2,3-二甲氧苯甲酸;5-磺酰胺基-2,3-二甲氧苯甲酸;2,3-二甲氧基-5-氨磺酰苯甲酸;2,3-二甲氧基-5-氨磺酰基苯甲酸;二甲氧基-5-磺酰胺基苯甲酸;2,3-二甲氧基-5-磺酰胺苯甲酸
英文名称
2,3-dimethoxy-5-sulphamoyl benzoic acid
英文别名
2,3-Dimethoxy-5-sulfamoylbenzoic Acid

CAS
66644-80-2
化学式
C9H11NO6S
mdl
MFCD01317561
分子量
261.255
InChiKey
YEVQOPOKMKTXMD-UHFFFAOYSA-N
BEILSTEIN
——
EINECS
——
-
物化性质
-
计算性质
-
ADMET
-
安全信息
-
SDS
-
制备方法与用途
-
上下游信息
-
文献信息
-
表征谱图
-
同类化合物
-
相关功能分类
-
相关结构分类
物化性质
-
熔点:206 °C
-
沸点:499.2±55.0 °C(Predicted)
-
密度:1.460±0.06 g/cm3(Predicted)
计算性质
-
辛醇/水分配系数(LogP):0
-
重原子数:17
-
可旋转键数:4
-
环数:1.0
-
sp3杂化的碳原子比例:0.222
-
拓扑面积:124
-
氢给体数:2
-
氢受体数:7
安全信息
-
危险品标志:Xn,Xi
-
WGK Germany:3
-
海关编码:29089000
-
安全说明:S26,S36,S36/37,S36/37/39
-
危险类别码:R20/21/22,R36/37/38
-
储存条件:室温且干燥
SDS
5-(氨基磺酰)-2,3-二甲氧苯甲酸
模块 1. 化学品
产品名称: 5-(Aminosulfonyl)-2,3-dimethoxybenzoic Acid
模块 2. 危险性概述
GHS分类
物理性危害 未分类
健康危害
皮肤腐蚀/刺激 第2级
严重损伤/刺激眼睛 2A类
环境危害 未分类
GHS标签元素
图标或危害标志
信号词 警告
危险描述 造成皮肤刺激
造成严重眼刺激
防范说明
[预防] 处理后要彻底清洗双手。
穿戴防护手套/护目镜/防护面具。
[急救措施] 眼睛接触:用水小心清洗几分钟。如果方便,易操作,摘除隐形眼镜。继续冲洗。
眼睛接触:求医/就诊
皮肤接触:用大量肥皂和水轻轻洗。
若皮肤刺激:求医/就诊。
脱掉被污染的衣物,清洗后方可重新使用。
模块 3. 成分/组成信息
单一物质/混和物 单一物质
化学名(中文名): 5-(氨基磺酰)-2,3-二甲氧苯甲酸
百分比: >98.0%(LC)(T)
CAS编码: 66644-80-2
俗名: 2,3-Dimethoxy-5-sulfamoylbenzoic Acid
5-(氨基磺酰)-2,3-二甲氧苯甲酸
模块 3. 成分/组成信息
分子式: C9H11NO6S
模块 4. 急救措施
吸入: 将受害者移到新鲜空气处,保持呼吸通畅,休息。若感不适请求医/就诊。
皮肤接触: 立即去除/脱掉所有被污染的衣物。用大量肥皂和水轻轻洗。
若皮肤刺激或发生皮疹:求医/就诊。
眼睛接触: 用水小心清洗几分钟。如果方便,易操作,摘除隐形眼镜。继续清洗。
如果眼睛刺激:求医/就诊。
食入: 若感不适,求医/就诊。漱口。
紧急救助者的防护: 救援者需要穿戴个人防护用品,比如橡胶手套和气密性护目镜。
模块 5. 消防措施
合适的灭火剂: 干粉,泡沫,雾状水,二氧化碳
特殊危险性: 小心,燃烧或高温下可能分解产生毒烟。
特定方法: 从上风处灭火,根据周围环境选择合适的灭火方法。
非相关人员应该撤离至安全地方。
周围一旦着火:如果安全,移去可移动容器。
消防员的特殊防护用具: 灭火时,一定要穿戴个人防护用品。
模块 6. 泄漏应急处理
个人防护措施,防护用具, 使用个人防护用品。远离溢出物/泄露处并处在上风处。
紧急措施: 泄露区应该用安全带等圈起来,控制非相关人员进入。
环保措施: 防止进入下水道。
控制和清洗的方法和材料: 清扫收集粉尘,封入密闭容器。注意切勿分散。附着物或收集物应该立即根据合适的
法律法规处置。
模块 7. 操作处置与储存
处理
技术措施: 在通风良好处进行处理。穿戴合适的防护用具。防止粉尘扩散。处理后彻底清洗双手
和脸。
注意事项: 如果粉尘或浮质产生,使用局部排气。
操作处置注意事项: 避免接触皮肤、眼睛和衣物。
贮存
储存条件: 保持容器密闭。存放于凉爽、阴暗处。
远离不相容的材料比如氧化剂存放。
包装材料: 依据法律。
模块 8. 接触控制和个体防护
工程控制: 尽可能安装封闭体系或局部排风系统,操作人员切勿直接接触。同时安装淋浴器和洗
眼器。
个人防护用品
呼吸系统防护: 防尘面具。依据当地和政府法规。
手部防护: 防护手套。
眼睛防护: 安全防护镜。如果情况需要,佩戴面具。
皮肤和身体防护: 防护服。如果情况需要,穿戴防护靴。
模块 9. 理化特性
外形(20°C): 固体
外观: 晶体-粉末
5-(氨基磺酰)-2,3-二甲氧苯甲酸
模块 9. 理化特性
颜色: 白色-极淡的黄色
气味: 无资料
pH: 无数据资料
熔点:
206°C
沸点/沸程 无资料
闪点: 无资料
爆炸特性
爆炸下限: 无资料
爆炸上限: 无资料
密度: 无资料
溶解度:
[水] 无资料
[其他溶剂] 无资料
模块 10. 稳定性和反应性
化学稳定性: 一般情况下稳定。
危险反应的可能性: 未报道特殊反应性。
须避免接触的物质 氧化剂
危险的分解产物: 一氧化碳, 二氧化碳, 氮氧化物 (NOx), 硫氧化物
模块 11. 毒理学信息
急性毒性: 无资料
对皮肤腐蚀或刺激: 无资料
对眼睛严重损害或刺激: 无资料
生殖细胞变异原性: 无资料
致癌性:
IARC = 无资料
NTP = 无资料
生殖毒性: 无资料
模块 12. 生态学信息
生态毒性:
鱼类: 无资料
甲壳类: 无资料
藻类: 无资料
残留性 / 降解性: 无资料
潜在生物累积 (BCF): 无资料
土壤中移动性
log水分配系数: 无资料
土壤吸收系数 (Koc): 无资料
亨利定律 无资料
constaNT(PaM3/mol):
模块 13. 废弃处置
如果可能,回收处理。请咨询当地管理部门。建议在可燃溶剂中溶解混合,在装有后燃和洗涤装置的化学焚烧炉中
焚烧。废弃处置时请遵守国家、地区和当地的所有法规。
模块 14. 运输信息
联合国分类: 与联合国分类标准不一致
5-(氨基磺酰)-2,3-二甲氧苯甲酸
模块 14. 运输信息
UN编号: 未列明
模块 15. 法规信息
《危险化学品安全管理条例》(2002年1月26日国务院发布,2011年2月16日修订): 针对危险化学品的安全使用、
生产、储存、运输、装卸等方面均作了相应的规定。
模块16 - 其他信息
N/A
模块 1. 化学品
产品名称: 5-(Aminosulfonyl)-2,3-dimethoxybenzoic Acid
模块 2. 危险性概述
GHS分类
物理性危害 未分类
健康危害
皮肤腐蚀/刺激 第2级
严重损伤/刺激眼睛 2A类
环境危害 未分类
GHS标签元素
图标或危害标志
信号词 警告
危险描述 造成皮肤刺激
造成严重眼刺激
防范说明
[预防] 处理后要彻底清洗双手。
穿戴防护手套/护目镜/防护面具。
[急救措施] 眼睛接触:用水小心清洗几分钟。如果方便,易操作,摘除隐形眼镜。继续冲洗。
眼睛接触:求医/就诊
皮肤接触:用大量肥皂和水轻轻洗。
若皮肤刺激:求医/就诊。
脱掉被污染的衣物,清洗后方可重新使用。
模块 3. 成分/组成信息
单一物质/混和物 单一物质
化学名(中文名): 5-(氨基磺酰)-2,3-二甲氧苯甲酸
百分比: >98.0%(LC)(T)
CAS编码: 66644-80-2
俗名: 2,3-Dimethoxy-5-sulfamoylbenzoic Acid
5-(氨基磺酰)-2,3-二甲氧苯甲酸
模块 3. 成分/组成信息
分子式: C9H11NO6S
模块 4. 急救措施
吸入: 将受害者移到新鲜空气处,保持呼吸通畅,休息。若感不适请求医/就诊。
皮肤接触: 立即去除/脱掉所有被污染的衣物。用大量肥皂和水轻轻洗。
若皮肤刺激或发生皮疹:求医/就诊。
眼睛接触: 用水小心清洗几分钟。如果方便,易操作,摘除隐形眼镜。继续清洗。
如果眼睛刺激:求医/就诊。
食入: 若感不适,求医/就诊。漱口。
紧急救助者的防护: 救援者需要穿戴个人防护用品,比如橡胶手套和气密性护目镜。
模块 5. 消防措施
合适的灭火剂: 干粉,泡沫,雾状水,二氧化碳
特殊危险性: 小心,燃烧或高温下可能分解产生毒烟。
特定方法: 从上风处灭火,根据周围环境选择合适的灭火方法。
非相关人员应该撤离至安全地方。
周围一旦着火:如果安全,移去可移动容器。
消防员的特殊防护用具: 灭火时,一定要穿戴个人防护用品。
模块 6. 泄漏应急处理
个人防护措施,防护用具, 使用个人防护用品。远离溢出物/泄露处并处在上风处。
紧急措施: 泄露区应该用安全带等圈起来,控制非相关人员进入。
环保措施: 防止进入下水道。
控制和清洗的方法和材料: 清扫收集粉尘,封入密闭容器。注意切勿分散。附着物或收集物应该立即根据合适的
法律法规处置。
模块 7. 操作处置与储存
处理
技术措施: 在通风良好处进行处理。穿戴合适的防护用具。防止粉尘扩散。处理后彻底清洗双手
和脸。
注意事项: 如果粉尘或浮质产生,使用局部排气。
操作处置注意事项: 避免接触皮肤、眼睛和衣物。
贮存
储存条件: 保持容器密闭。存放于凉爽、阴暗处。
远离不相容的材料比如氧化剂存放。
包装材料: 依据法律。
模块 8. 接触控制和个体防护
工程控制: 尽可能安装封闭体系或局部排风系统,操作人员切勿直接接触。同时安装淋浴器和洗
眼器。
个人防护用品
呼吸系统防护: 防尘面具。依据当地和政府法规。
手部防护: 防护手套。
眼睛防护: 安全防护镜。如果情况需要,佩戴面具。
皮肤和身体防护: 防护服。如果情况需要,穿戴防护靴。
模块 9. 理化特性
外形(20°C): 固体
外观: 晶体-粉末
5-(氨基磺酰)-2,3-二甲氧苯甲酸
模块 9. 理化特性
颜色: 白色-极淡的黄色
气味: 无资料
pH: 无数据资料
熔点:
206°C
沸点/沸程 无资料
闪点: 无资料
爆炸特性
爆炸下限: 无资料
爆炸上限: 无资料
密度: 无资料
溶解度:
[水] 无资料
[其他溶剂] 无资料
模块 10. 稳定性和反应性
化学稳定性: 一般情况下稳定。
危险反应的可能性: 未报道特殊反应性。
须避免接触的物质 氧化剂
危险的分解产物: 一氧化碳, 二氧化碳, 氮氧化物 (NOx), 硫氧化物
模块 11. 毒理学信息
急性毒性: 无资料
对皮肤腐蚀或刺激: 无资料
对眼睛严重损害或刺激: 无资料
生殖细胞变异原性: 无资料
致癌性:
IARC = 无资料
NTP = 无资料
生殖毒性: 无资料
模块 12. 生态学信息
生态毒性:
鱼类: 无资料
甲壳类: 无资料
藻类: 无资料
残留性 / 降解性: 无资料
潜在生物累积 (BCF): 无资料
土壤中移动性
log水分配系数: 无资料
土壤吸收系数 (Koc): 无资料
亨利定律 无资料
constaNT(PaM3/mol):
模块 13. 废弃处置
如果可能,回收处理。请咨询当地管理部门。建议在可燃溶剂中溶解混合,在装有后燃和洗涤装置的化学焚烧炉中
焚烧。废弃处置时请遵守国家、地区和当地的所有法规。
模块 14. 运输信息
联合国分类: 与联合国分类标准不一致
5-(氨基磺酰)-2,3-二甲氧苯甲酸
模块 14. 运输信息
UN编号: 未列明
模块 15. 法规信息
《危险化学品安全管理条例》(2002年1月26日国务院发布,2011年2月16日修订): 针对危险化学品的安全使用、
生产、储存、运输、装卸等方面均作了相应的规定。
模块16 - 其他信息
N/A
上下游信息
-
上游原料
中文名称 英文名称 CAS号 化学式 分子量 2,3-二甲氧基-5-氨基磺酰基苯甲酸甲酯 2,3-dimethoxy-5-sulfonamidobenzoic acid methyl ester 66644-82-4 C10H13NO6S 275.282 3,4-二甲氧基苯磺酰胺 3,4-dimethoxybenzenesulfonamide 63624-27-1 C8H11NO4S 217.246 -
下游产品
中文名称 英文名称 CAS号 化学式 分子量 5-(氨基磺酰基)-2,3-二甲氧基苯甲酰氯 2,3-dimethoxy-5-sulphamoyl benzoyl chloride 57734-47-1 C9H10ClNO5S 279.701 维拉必利 Veralipride 66644-81-3 C17H25N3O5S 383.469 —— N-(1-cycloheptylmethyl-pyrrolidin-2-ylmethyl)-2,3-dimethoxy-5-sulfamoyl-benzamide 72135-89-8 C22H35N3O5S 453.603 —— N-[1-(cyclohexenyl)-methyl-2-pyrrolidylmethyl]-2,3-dimethoxy-5-sulphamoyl-benzamide 72135-14-9 C21H31N3O5S 437.56
反应信息
-
作为反应物:描述:参考文献:名称:New substituted heterocyclic benzamides, methods of preparing them and摘要:这里提供了新的替代杂环苯甲酰胺及其衍生物,这些化合物对中枢神经系统产生改变。公开号:US04673686A1
-
作为产物:描述:3,4-二甲氧基苯磺酰胺 在 盐酸 、 copper(l) iodide 、 水 、 溴 、 铁粉 、 sodium hydroxide 作用下, 以 二氯甲烷 、 N,N-二甲基甲酰胺 为溶剂, 反应 44.0h, 生成 2,3-二甲氧基-5-磺酰胺基苯甲酸参考文献:名称:一种2,3-二甲氧基-5-磺酰胺基苯甲酸的制备新方法摘要:一种2,3‑二甲氧基‑5‑磺酰胺基苯甲酸的制备新方法,涉及一种医药中间体的制备方法。该方法依次包括如下步骤:采用3,4‑二羟基苯磺酰胺为原料,依次与溴化和取代反应,最后在两次水解下得到产品2,3‑二甲氧基‑5‑磺酰胺基苯甲酸,本发明提出了一条全新的合成路线,所用原料来源广泛充足,价格便宜、反应条件温和、工艺简洁,各步反应均为常规操作,避免了使用剧毒的硫酸二甲酯等有害原料,并降低了污染,有较好的应用范围。公开号:CN111018750B
文献信息
-
[DE] STEROID-PRODRUGS MIT ANDROGENER WIRKUNG<br/>[EN] STEROID PRODRUGS WITH ANDROGENIC EFFECT<br/>[FR] PROMEDICAMENTS STEROIDES A EFFET ANDROGENE申请人:SCHERING AG公开号:WO2005113575A1公开(公告)日:2005-12-01Zusammenfassung Die vorliegende Erfindung betrifft Steroid-Prodrugs mit androgener Wirkung der allgemeinen Formel (I), in denen die Gruppe Z an das Steroid gebunden ist, pharmazeutische Zusammensetzungen, die diese Verbindungen enthalten sowie deren Verwendung zur Herstellung von Arzneimitteln mit androgener Wirkung.
-
Synthesis and in silico evaluation of novel uridyl sulfamoylbenzoate derivatives as potential anticancer agents targeting M1 subunit of human ribonucleotide reductase (hRRM1)作者:Prince J. Salvador、Heather B. Jacobs、Lujain Alnouri、Asia Fee、Lynn M. Utley、Madison Mabry、Hannah Krajeck、Christopher Dicksion、Ahmed M. AwadDOI:10.1007/s00044-021-02840-4日期:2022.7novo synthesis of deoxynucleotide triphosphates (dNTPs). Nucleoside analogues have been investigated as anticancer drugs that inhibit human RNR, however, problems with toxicity and cancer resistance remain challenging. Herein we report a convenient synthesis of six novel nucleoside analogues modified with benzenesulfonamide derivatives: 4-carboxybenzenesulfonamide, 4-chloro-3-sulfamoylbenzoic acid, 2核糖核苷酸还原酶(RNR)是癌症化疗的关键靶点。该酶催化核糖核苷酸还原为 2'-脱氧核糖核苷酸,其活性在脱氧核苷酸三磷酸 (dNTP) 的从头合成中是限速的。核苷类似物已被研究作为抑制人类 RNR 的抗癌药物,然而,毒性和抗癌性问题仍然具有挑战性。在这里,我们报告了六种用苯磺酰胺衍生物修饰的新型核苷类似物的简便合成方法:4-羧基苯磺酰胺、4-氯代-3-氨磺酰苯甲酸、2-氯代-4-氟代-5-氨磺酰苯甲酸、2,3-二甲氧基-5-氨磺酰基苯甲酸,N-苄基-4-氯-5-氨磺酰邻氨基苯甲酸,或速尿。苯甲酸磺酰胺的羧基与尿苷的 5' 羟基之间的 Mitsunobu 反应以优异的产率产生了尿嘧啶基氨磺酰基苯甲酸酯。进行分子对接以检查与 RNR 的大亚基 M1 的构象和结合亲和力。氨磺酰基部分与催化位点中已知的底物结合残基如 Ser202 和 Thr607 显示出强氢键。吸电子氟和氯增强了结合,而给电子甲氧基降低了结合。计算机
-
[DE] AMINOSULFONYL- ODER AMINOSULFONYLAMINO-SUBSTITUIERTE PHENYL-ESTER ALS ESTRADIOL-PRODRUGS<br/>[EN] AMINOSULPHONYL- OR AMINOSULPHONYLAMINO-SUBSTITUTED PHENYL ESTERS AS ESTRADIOL PRODRUGS<br/>[FR] PHENYLESTERS SUBSTITUES PAR AMINOSULFONYLE OU AMINOSULFONYLAMINO COMME PROMEDICAMENTS A BASE D'OESTRADIOL申请人:SCHERING AG公开号:WO2005113574A1公开(公告)日:2005-12-01Die Erfindung betrifft Estradiol-Prodrugs der allgemeinen Formel (I), in denen die Gruppe Z an das Steroid gebunden ist, Verfahren zu deren Herstellung, pharmazeutische Zusammensetzungen, die diese Verbindungen enthalten sowie deren Verwendung zur Herstellung von Arzneimitteln mit estrogener Wirkung.
-
Estradiol prodrugs申请人:Wyrwa Ralf公开号:US20050288267A1公开(公告)日:2005-12-29The invention relates to estradiol prodrugs of general formula (I), in which group Z is bonded to the steroid, process for their production, pharmaceutical compositions that contain these compounds, as well as their use for the production of pharmaceutical agents with estrogenic action.
-
N-(1-allyl-2-pyrrolidylmethyl)-2,3-dimethoxy-5-sulfamoylbenzamide and申请人:Societe D'Etudes Scientifiques et Industrielles de l'Ile de France公开号:US04499019A1公开(公告)日:1985-02-12N-(1'-allyl-2'-pyrrolidylmethyl)-2,3-dimethoxy-5-sulfamoylbenzamide and dvatives thereof and method for treating the psycho neuro-vegetative syndrome of the natural or surgical menopause by administering an effective amount of said compound to a patient.
表征谱图
-
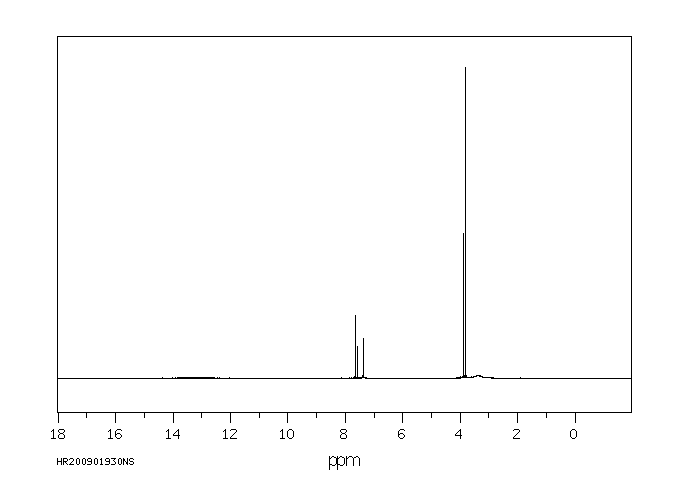
氢谱1HNMR
-
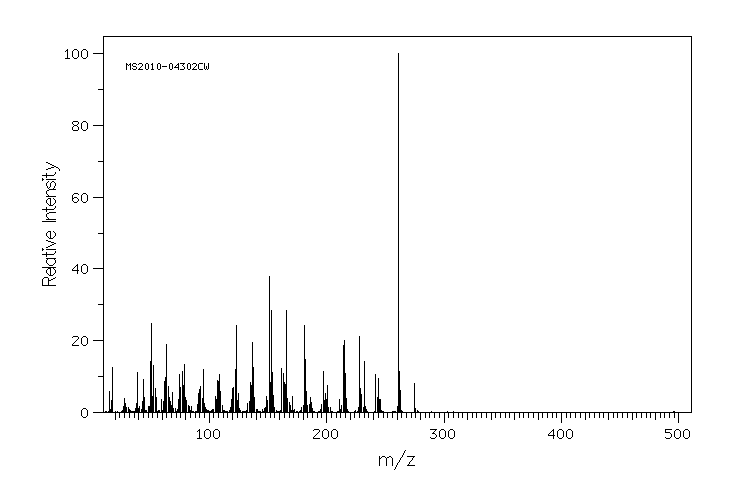
质谱MS
-
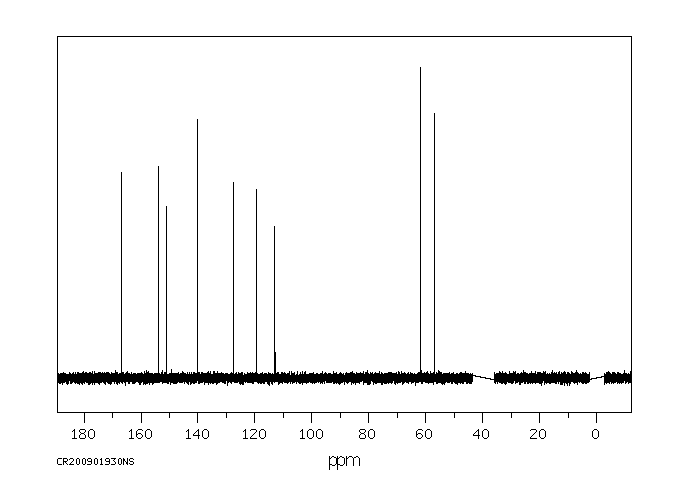
碳谱13CNMR
-
红外IR
-
拉曼Raman
-
峰位数据
-
峰位匹配
-
表征信息
同类化合物
(βS)-β-氨基-4-(4-羟基苯氧基)-3,5-二碘苯甲丙醇
(S,S)-邻甲苯基-DIPAMP
(S)-(-)-7'-〔4(S)-(苄基)恶唑-2-基]-7-二(3,5-二-叔丁基苯基)膦基-2,2',3,3'-四氢-1,1-螺二氢茚
(S)-盐酸沙丁胺醇
(S)-3-(叔丁基)-4-(2,6-二甲氧基苯基)-2,3-二氢苯并[d][1,3]氧磷杂环戊二烯
(S)-2,2'-双[双(3,5-三氟甲基苯基)膦基]-4,4',6,6'-四甲氧基联苯
(S)-1-[3,5-双(三氟甲基)苯基]-3-[1-(二甲基氨基)-3-甲基丁烷-2-基]硫脲
(R)富马酸托特罗定
(R)-(-)-盐酸尼古地平
(R)-(-)-4,12-双(二苯基膦基)[2.2]对环芳烷(1,5环辛二烯)铑(I)四氟硼酸盐
(R)-(+)-7-双(3,5-二叔丁基苯基)膦基7''-[((6-甲基吡啶-2-基甲基)氨基]-2,2'',3,3''-四氢-1,1''-螺双茚满
(R)-(+)-7-双(3,5-二叔丁基苯基)膦基7''-[(4-叔丁基吡啶-2-基甲基)氨基]-2,2'',3,3''-四氢-1,1''-螺双茚满
(R)-(+)-7-双(3,5-二叔丁基苯基)膦基7''-[(3-甲基吡啶-2-基甲基)氨基]-2,2'',3,3''-四氢-1,1''-螺双茚满
(R)-(+)-4,7-双(3,5-二-叔丁基苯基)膦基-7“-[(吡啶-2-基甲基)氨基]-2,2”,3,3'-四氢1,1'-螺二茚满
(R)-3-(叔丁基)-4-(2,6-二苯氧基苯基)-2,3-二氢苯并[d][1,3]氧杂磷杂环戊烯
(R)-2-[((二苯基膦基)甲基]吡咯烷
(R)-1-[3,5-双(三氟甲基)苯基]-3-[1-(二甲基氨基)-3-甲基丁烷-2-基]硫脲
(N-(4-甲氧基苯基)-N-甲基-3-(1-哌啶基)丙-2-烯酰胺)
(5-溴-2-羟基苯基)-4-氯苯甲酮
(5-溴-2-氯苯基)(4-羟基苯基)甲酮
(5-氧代-3-苯基-2,5-二氢-1,2,3,4-oxatriazol-3-鎓)
(4S,5R)-4-甲基-5-苯基-1,2,3-氧代噻唑烷-2,2-二氧化物-3-羧酸叔丁酯
(4S,4''S)-2,2''-亚环戊基双[4,5-二氢-4-(苯甲基)恶唑]
(4-溴苯基)-[2-氟-4-[6-[甲基(丙-2-烯基)氨基]己氧基]苯基]甲酮
(4-丁氧基苯甲基)三苯基溴化磷
(3aR,8aR)-(-)-4,4,8,8-四(3,5-二甲基苯基)四氢-2,2-二甲基-6-苯基-1,3-二氧戊环[4,5-e]二恶唑磷
(3aR,6aS)-5-氧代六氢环戊基[c]吡咯-2(1H)-羧酸酯
(2Z)-3-[[(4-氯苯基)氨基]-2-氰基丙烯酸乙酯
(2S,3S,5S)-5-(叔丁氧基甲酰氨基)-2-(N-5-噻唑基-甲氧羰基)氨基-1,6-二苯基-3-羟基己烷
(2S,2''S,3S,3''S)-3,3''-二叔丁基-4,4''-双(2,6-二甲氧基苯基)-2,2'',3,3''-四氢-2,2''-联苯并[d][1,3]氧杂磷杂戊环
(2S)-(-)-2-{[[[[3,5-双(氟代甲基)苯基]氨基]硫代甲基]氨基}-N-(二苯基甲基)-N,3,3-三甲基丁酰胺
(2S)-2-[[[[[((1S,2S)-2-氨基环己基]氨基]硫代甲基]氨基]-N-(二苯甲基)-N,3,3-三甲基丁酰胺
(2S)-2-[[[[[[((1R,2R)-2-氨基环己基]氨基]硫代甲基]氨基]-N-(二苯甲基)-N,3,3-三甲基丁酰胺
(2-硝基苯基)磷酸三酰胺
(2,6-二氯苯基)乙酰氯
(2,3-二甲氧基-5-甲基苯基)硼酸
(1S,2S,3S,5S)-5-叠氮基-3-(苯基甲氧基)-2-[(苯基甲氧基)甲基]环戊醇
(1S,2S,3R,5R)-2-(苄氧基)甲基-6-氧杂双环[3.1.0]己-3-醇
(1-(4-氟苯基)环丙基)甲胺盐酸盐
(1-(3-溴苯基)环丁基)甲胺盐酸盐
(1-(2-氯苯基)环丁基)甲胺盐酸盐
(1-(2-氟苯基)环丙基)甲胺盐酸盐
(1-(2,6-二氟苯基)环丙基)甲胺盐酸盐
(-)-去甲基西布曲明
龙蒿油
龙胆酸钠
龙胆酸叔丁酯
龙胆酸
龙胆紫-d6
龙胆紫
相关功能分类

联系我们



关注我们

公众号

