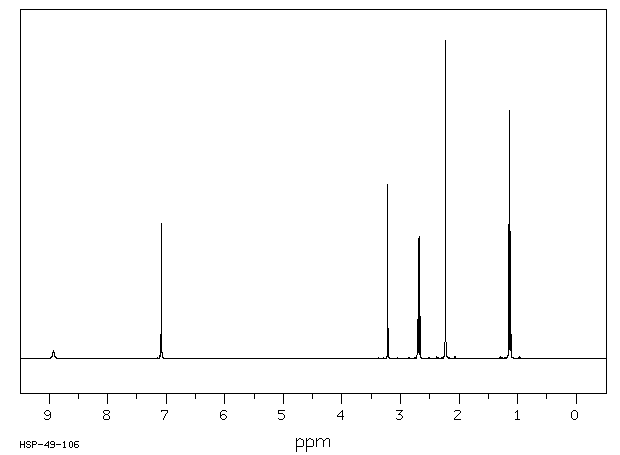
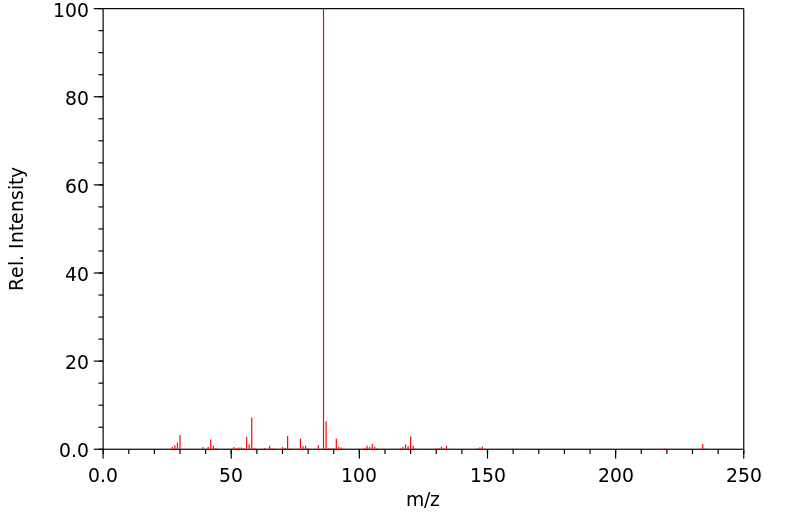
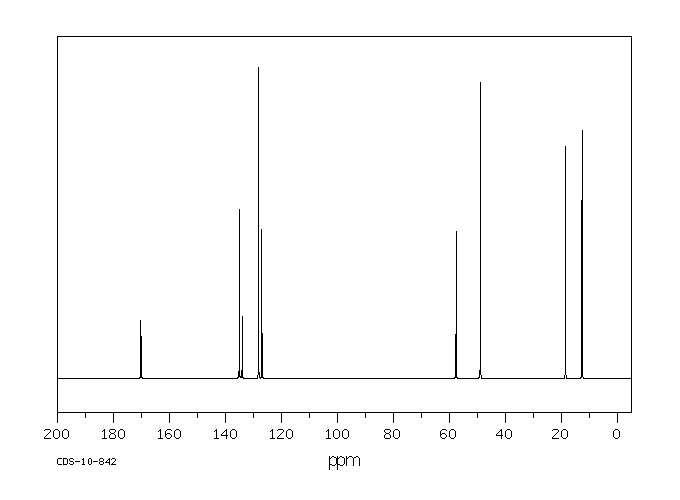
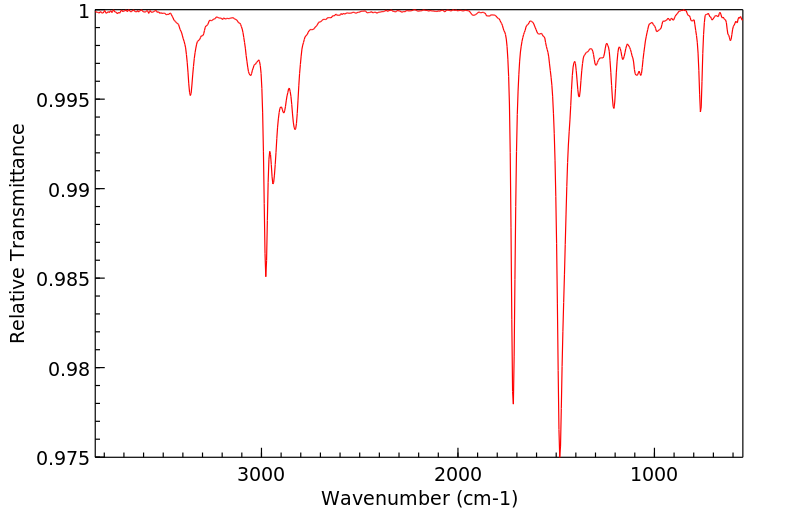
Commercially available solutions of lidocaine hydrochloride in 5% dextrose usually are stable for 18 months after the date of manufacture. Commercially available solutions of lidocaine hydrochloride in 5% dextrose may be provided in plastic containers.
分解:
When heated to decomposition it emits toxic fumes of /nitrogen oxides/.
Lidocaine is metabolized predominantly and rapidly by the liver, and metabolites and unchanged drug are excreted by the kidneys. Biotransformation includes oxidative N-dealkylation, ring hydroxylation, cleavage of the amide linkage, and conjugation. N-dealkylation, a major pathway of biotransformation, yields the metabolites monoethylglycinexylidide and glycinexylidide. The pharmacological/toxicological actions of these metabolites are similar to, but less potent than, those of lidocaine HCl. Approximately 90% of lidocaine HCl administered is excreted in the form of various metabolites, and less than 10% is excreted unchanged. The primary metabolite in urine is a conjugate of 4-hydroxy-2,6-dimethylaniline.
Approximately 90% of a parenteral dose of lidocaine is rapidly metabolized in the liver by de-ethylation to form MEGX and GX followed by cleavage of the amide bond to form xylidine and 4-hydroxyxylidine which are excreted in urine. Less than 10% of a dose is excreted unchanged in urine.
The rate of lidocaine metabolism may also be decreased in patients with liver disease, possibly because of altered perfusion in the liver or hepatic tissue necrosis. Distribution and elimination of lidocaine and /monoethylglycinexylidide/ MEGX appear to remain normal in patients with renal failure, but /glycinexylidide/ GX may accumulate in these patients when lidocaine is administered IV for several days.
... The purpose of this study is to determine the amount of lidocaine and its metabolite monoethyl-glycinexylidide (MEGX) in breast milk after local anesthesia during dental procedures. The study population consisted of seven nursing mothers (age, 23-39 years) who received 3.6 to 7.2 mL 2% lidocaine without adrenaline. Blood and milk concentrations of lidocaine and its metabolite MEGX were assayed using high-performance liquid chromatography. The milk-to-plasma ratio and the possible daily doses in infants for both lidocaine and MEGX were calculated. The lidocaine concentration in maternal plasma 2 hours after injection was 347.6 +/- 221.8 ug/L, the lidocaine concentration in maternal milk ranged from 120.5 +/- 54.1 ug/L (3 hours after injection) to 58.3 +/- 22.8 ug/L (6 hours after injection), the MEGX concentration in maternal plasma 2 hours after injection was 58.9 +/- 30.3 ug/L, and the MEGX concentration in maternal milk ranged from 97.5 +/- 39.6 ug/L (3 hours after injection) to 52.7 +/- 23.8 ug/L (6 hours after injection). According to these data and considering an intake of 90 mL breast milk every 3 hours, the daily infant dosages of lidocaine and MEGX were 73.41 +/- 38.94 ug/L/day and 66.1 +/- 28.5 ug/L/day respectively. This study suggests that even if a nursing mother undergoes dental treatment with local anesthesia using lidocaine without adrenaline, she can safely continue breastfeeding.
... To determine the time/concentration profile of lidocaine and its active metabolites glycinexylidide (GX) and monoethylglycinexylidide (MEGX) during a 96 hr lidocaine infusion. lidocaine was administered to 8 mature healthy horses as a continuous rate infusion (0.05 mg/kg bwt/min) for 96 hr. Blood concentrations of lidocaine, GX and MEGX were determined using high performance liquid chromatography during and after discontinuation of the infusion. Serum lidocaine concentrations reached steady state by 3 hr and did not accumulate thereafter. Concentrations were above the target therapeutic concentration (980 ng/mL) only at 6 and 48 hr, and did not reach the range described as potentially causing toxicity (>1850 ng/mL) at any time. MEGX did not accumulate over time, while the GX accumulated significantly up to 48 hr and then remained constant. The serum concentrations of lidocaine, MEGX and GX were below the limit of detection within 24 hr of discontinuation of the infusion. None of the horses developed any signs of lidocaine toxicity during the study. The metabolism of lidocaine was not significantly impaired by prolonged infusion and no adverse effects were observed. Prolonged infusions appear to be safe in normal horses but the accumulation of GX, a potentially toxic active metabolite, is cause for concern.
IDENTIFICATION AND USE: Lidocaine is a white or slightly yellow, crystalline powder or needle with a characteristic odor. It is commonly used as a medication including for local anesthetics, anti-arrhythmia agent, or as a voltage-gated sodium channel blocker. Lidocaine may also be used in the treatment of hypertensive emergencies, or acute coronary syndrome associated with the toxicity of various stimulants and antiarrhythmic agents. A lidocaine transdermal patch is used for relief of pain associated with postherpetic neuralgia. An oral patch is available for application to accessible mucous membranes of the mouth prior to superficial dental procedures. The combination of lidocaine (2.5%) and prilocaine (2.5%) in an occlusive dressing is used as an anesthetic prior to venipuncture, skin graft harvesting, and infiltration of anesthetics into genitalia. Lidocaine in combination with tetracaine in a formulation that generates a "peel" is approved for topical local analgesia prior to superficial dermatological procedures. HUMAN EXPOSURE AND TOXICITY: Adverse effects of the drug mainly involve the CNS because of its rapid entry in the brain. Adverse CNS reactions may be manifested by drowsiness; dizziness; disorientation; confusion; lightheadedness; tremulousness; psychosis; nervousness; apprehension; agitation; euphoria; tinnitus; visual disturbances including blurred or double vision; nausea; vomiting; paresthesia; sensations of neat, cold or numbness; difficulty swallowing; dyspnea; and slurred speech. Muscle twitching or tremors, seizures, unconsciousness, coma, and respiratory depression and arrest may also occur. Shortly following the CNS effects, patients with lidocaine toxicity may also experience cardiovascular effects. If the patient is supported through this period, the drug rapidly distributes away from the heart, and spontaneous cardiac function returns. Lidocaine, when administered to a baby may induce convulsions. Lidocaine intoxication in the neonate, occurring as a result of inadvertent injection into the fetal scalp or cranium during local anesthesia (caudal or paracervical block or episiotomy), produces apnea, hypotonia, and seizures. Dilated pupils and loss of the oculocephalic reflex may also be observed. The more severe of these effects develop when serum lidocaine concentrations exceed 5 ug/mL and are often preceded by paresthesias or somnolence. Continuous application for 72 hours of four lidocaine patches 5%, changed every 12 or 24 hours, produced mild application-site erythema in most patients, but no systemic adverse reactions. No loss in sensation at the application site was reported. Systemic exposure to lidocaine and monoethylglycinexylidide (MEGX), the primary active metabolite of lidocaine, after application of lidocaine gel or patches was minimal in normal volunteers, patients with post-herpetic neuralgia, and patients with acute herpes zoster. In human SH-SY5Y neuroblastoma cells, local anesthesia caused rapid cell death, which was primarily due to necrosis. Lidocaine can trigger apoptosis with either increased time of exposure or increased concentration. ANIMAL STUDIES: In rats persistent functional impairment and histologic damage in the nerve roots and the spinal cord was less severe after epidural lidocaine than after intrathecal lidocaine. In 8 New Zealand Rabbits receiving 0.2 mL 1% lidocaine hydrochloride applied intracamerally to the lenses, had morphological abnormalities in both cornea and iris of the lidocaine injected eyes. Another experiment in rabbits with 2% lidocaine HCl applied intracamerally on the corneal endothelium found that lidocaine caused statistically significant corneal thickening and clinically significant corneal opacification. Lidocaine injection into the dorsal root ganglion of rats produced hyperalgesia, possibly due to activation of resident satellite glial cells. One-hour exposure of primary rabbit urothelial cells (PRUC) culture to 0.5 or 1.0% lidocaine decreased cell viability. Lidocaine rapidly crosses the placenta in pregnant guinea pigs. High concentrations are found in the fetal liver, heart, and brain. High myocardial levels of drug in the fetus may possibly account for marked depressant effects that local anesthetics produce. In another study, no significant effects were observed in offspring of rats administered lidocaine at by constant infusion for 2 weeks before mating and throughout pregnancy. Additionally, pregnancy did not enhance the CNS and cardiovascular toxic effects of lidocaine when studied in pregnant sheep receiving continuous IV drug infusion and compared to data from nonpregnant ewes. Lidocaine did not induce genotoxicity in the wing somatic mutation and recombination test in Drosophila melanogaster, which detects point and chromosomal mutations as well as recombination induced by the activity of genotoxins of direct and indirect action. Lidocaine 0.25% did decrease cell viability and caused DNA degradation in murine fibroblasts 3T6. Lidocaine was not oncogenic when administered topically weekly to the dorsal skin of mice for 26 weeks.
Lidocaine stabilizes the neuronal membrane by inhibiting the ionic fluxes required for the initiation and conduction of impulses thereby effecting local anesthetic action. Lidocaine alters signal conduction in neurons by blocking the fast voltage gated sodium (Na+) channels in the neuronal cell membrane that are responsible for signal propagation. With sufficient blockage the membrane of the postsynaptic neuron will not depolarize and will thus fail to transmit an action potential. This creates the anaesthetic effect by not merely preventing pain signals from propagating to the brain but by aborting their birth in the first place.
In general, lidocaine is readily absorbed across mucous membranes and damaged skin but poorly through intact skin. The agent is quickly absorbed from the upper airway, tracheobronchial tree, and alveoli into the bloodstream. And although lidocaine is also well absorbed across the gastrointestinal tract the oral bioavailability is only about 35% as a result of a high degree of first-pass metabolism. After injection into tissues, lidocaine is also rapidly absorbed and the absorption rate is affected by both vascularity and the presence of tissue and fat capable of binding lidocaine in the particular tissues. The concentration of lidocaine in the blood is subsequently affected by a variety of aspects, including its rate of absorption from the site of injection, the rate of tissue distribution, and the rate of metabolism and excretion. Subsequently, the systemic absorption of lidocaine is determined by the site of injection, the dosage given, and its pharmacological profile. The maximum blood concentration occurs following intercostal nerve blockade followed in order of decreasing concentration, the lumbar epidural space, brachial plexus site, and subcutaneous tissue. The total dose injected regardless of the site is the primary determinant of the absorption rate and blood levels achieved. There is a linear relationship between the amount of lidocaine injected and the resultant peak anesthetic blood levels. Nevertheless, it has been observed that lidocaine hydrochloride is completely absorbed following parenteral administration, its rate of absorption depending also on lipid solubility and the presence or absence of a vasoconstrictor agent. Except for intravascular administration, the highest blood levels are obtained following intercostal nerve block and the lowest after subcutaneous administration. Additionally, lidocaine crosses the blood-brain and placental barriers, presumably by passive diffusion.
The excretion of unchanged lidocaine and its metabolites occurs predominantly via the kidney with less than 5% in the unchanged form appearing in the urine. The renal clearance is inversely related to its protein binding affinity and the pH of the urine. This suggests by the latter that excretion of lidocaine occurs by non-ionic diffusion.
The volume of distribution determined for lidocaine is 0.7 to 1.5 L/kg. In particular, lidocaine is distributed throughout the total body water. Its rate of disappearance from the blood can be described by a two or possibly even three-compartment model. There is a rapid disappearance (alpha phase) which is believed to be related to uptake by rapidly equilibrating tissues (tissues with high vascular perfusion, for example). The slower phase is related to distribution to slowly equilibrating tissues (beta phase) and to its metabolism and excretion (gamma phase). Lidocaine's distribution is ultimately throughout all body tissues. In general, the more highly perfused organs will show higher concentrations of the agent. The highest percentage of this drug will be found in skeletal muscle, mainly due to the mass of muscle rather than an affinity.
Binding of lidocaine to plasma proteins is variable and concentration dependent. At concentrations of 1-4 ug/mL, the drug is approximately 60-80% bound to plasma proteins. Lidocaine is partially bound to a1-acid glycoprotein (a1-AGP), and the extent of binding to a1-AGP depends on the plasma concentration of the protein. In patients with myocardial infarction, increases in plasma a1-AGP concentration are associated with increased lidocaine binding and increased total plasma concentrations of the drug, but only small increases in plasma concentration of free drug; these changes in a1-AGP concentration and lidocaine binding are believed to account in part for accumulation of the drug observed in patients with myocardial infarction receiving prolonged infusions.
[EN] SUBSTITUTED N-HETEROCYCLIC CARBOXAMIDES AS ACID CERAMIDASE INHIBITORS AND THEIR USE AS MEDICAMENTS<br/>[FR] CARBOXAMIDES N-HÉTÉROCYCLIQUES SUBSTITUÉS UTILISÉS EN TANT QU'INHIBITEURS DE LA CÉRAMIDASE ACIDE ET LEUR UTILISATION EN TANT QUE MÉDICAMENTS
申请人:BIAL BIOTECH INVEST INC
公开号:WO2021055627A1
公开(公告)日:2021-03-25
The invention provides substituted N-heterocyclic carboxamides and related compounds, compositions containing such compounds, medical kits, and methods for using such compounds and compositions to treat a medical disorder, e.g., cancer, lysosomal storage disorder, neurodegenerative disorder, inflammatory disorder, in a patient.
Eflornithine Prodrugs, Conjugates and Salts, and Methods of Use Thereof
申请人:Xu Feng
公开号:US20100120727A1
公开(公告)日:2010-05-13
In one aspect, the present invention provides a composition of a covalent conjugate of an eflornithine analog with an anti-inflammatory drug. In another aspect, the present invention provides a composition of an eflornithine prodrug. In another aspect, the present invention provides a composition of an eflornithine or its derivatives aspirin salt. In another aspect, the present invention provides methods for treating or preventing cancer using the conjugates or salts of eflornithine analogs or eflornithine prodrugs.
[EN] THIENOPYRIDONE DERIVATIVES AS AMP-ACTIVATED PROTEIN KINASE (AMPK) ACTIVATORS<br/>[FR] DÉRIVÉS DE THÉNOPYRIDONE COMME ACTIVATEURS DE LA PROTÉINE KINASE DÉPENDANTE DE L'AMP (AMPK)
申请人:MERCK PATENT GMBH
公开号:WO2009124636A1
公开(公告)日:2009-10-15
The present invention relates to compounds of formula (I) wherein R1, R2 and R3 are as defined in claim 1, including pharmaceutical compositions thereof and for their use in the treatment and/or prevention of diseases and disorders modulated by AMP agonists. The invention is also directed to intermediates and to a method of preparation of compounds of formula (I).
THIENOPYRIDONE DERIVATIVES AS AMP-ACTIVATED PROTEIN KINASE (AMPK) ACTIVATORS
申请人:Cravo Daniel
公开号:US20110034505A1
公开(公告)日:2011-02-10
The present invention relates to compounds of formula (I) wherein R1, R2 and R3 are as defined in claim
1
, including pharmaceutical compositions thereof and for their use in the treatment and/or prevention of diseases and disorders modulated by AMP agonists. The invention is also directed to intermediates and to a method of preparation of compounds of formula (I).
[EN] ARYL ETHER-BASE KINASE INHIBITORS<br/>[FR] INHIBITEURS DE KINASES DE TYPE ARYLÉTHER-BASE
申请人:BRISTOL MYERS SQUIBB CO
公开号:WO2015038112A1
公开(公告)日:2015-03-19
The present disclosure is generally directed to compounds which can inhibit AAK1 (adaptor associated kinase 1), compositions comprising such compounds, and methods for inhibiting AAK1.