
噻唑烷-2-甲酸 | 16310-13-7
物质功能分类
中文名称
噻唑烷-2-甲酸
中文别名
噻唑烷-2-甲酸;四氢噻唑-2-甲酸;噻唑啉-2-甲酸;1,3-噻唑烷-2-甲酸;四氢噻唑-2-甲酸;噻唑烷-2-甲酸:四氢噻唑-2-甲酸; 噻唑啉-2-甲酸
英文名称
DL-3-thiazolidine-4-carboxylic acid
英文别名
thiazolidine-2-Carboxylic acid;1,3-thiazolidin-3-ium-2-carboxylate

CAS
16310-13-7
化学式
C4H7NO2S
mdl
MFCD00068005
分子量
133.171
InChiKey
ULSZVNJBVJWEJE-UHFFFAOYSA-N
BEILSTEIN
——
EINECS
——
-
物化性质
-
计算性质
-
ADMET
-
安全信息
-
SDS
-
制备方法与用途
-
上下游信息
-
文献信息
-
表征谱图
-
同类化合物
-
相关功能分类
-
相关结构分类
物化性质
-
熔点:179-184 °C
-
沸点:350.3±37.0 °C(Predicted)
-
密度:1.380±0.06 g/cm3(Predicted)
-
稳定性/保质期:
常温常压下稳定,避免与氧化物接触。
计算性质
-
辛醇/水分配系数(LogP):-2.3
-
重原子数:8
-
可旋转键数:1
-
环数:1.0
-
sp3杂化的碳原子比例:0.75
-
拓扑面积:74.6
-
氢给体数:2
-
氢受体数:4
安全信息
-
危险品标志:Xn
-
安全说明:S26,S36/37/39
-
危险类别码:R20/21/22,R36/37/38
-
海关编码:2934999090
-
储存条件:常温下应密闭避光保存,并保持通风和干燥。
SDS
制备方法与用途
噻唑烷-2-甲酸可用作有机合成中间体和医药中间体,主要应用于实验室研发及医药化工生产过程。
上下游信息
反应信息
-
作为反应物:参考文献:名称:Substituted n-(indole-2-carbonyl)-glycinamides and derivatives as摘要:公式(1)的化合物,其中R6是羧基,(C1-C8)烷氧基羰基,苄氧基羰基,C(O)NR8R9或C(O)R12作为葡萄糖磷酸化酶抑制剂,包含此类抑制剂的药物组合物以及使用此类抑制剂治疗哺乳动物中的糖尿病、高血糖、高胆固醇血症、高血压、高胰岛素血症、高脂血症、动脉粥样硬化和心肌缺血。公开号:US06107329A1
-
作为产物:描述:参考文献:名称:Pasenko, Ukrainskij Khimicheskij Zhurnal, 1958, vol. 24, p. 632,634摘要:DOI:
文献信息
-
Synthesis of novel thiazolothiazepine based HIV-1 integrase inhibitors作者:Francesca Aiello、Antonella Brizzi、Antonio Garofalo、Fedora Grande、Gaetano Ragno、Raveendra Dayam、Nouri NeamatiDOI:10.1016/j.bmc.2004.05.037日期:2004.8smallest and most constrained inhibitors of human immunodeficiency virus type-1 integrase (HIV-1 IN) inhibitors (J. Med. Chem. 1999, 42, 3334). Previously, we identified two thiazolothiazepines lead IN inhibitors with antiviral activity in cell-based assays. Structural optimization of these molecules necessitated the design of easily synthesizable analogs. In order to design similar molecules with least噻唑并氮杂卓类是人类免疫缺陷病毒1型整合酶(HIV-1 IN)抑制剂的最小和最受限制的抑制剂之一(J. Med。Chem。1999,42,3334)。以前,我们在基于细胞的测定法中鉴定了两种具有抗病毒活性的噻唑并噻嗪类铅IN抑制剂。这些分子的结构优化需要设计易于合成的类似物。为了设计具有最少数量取代基的相似分子,本文报道了10种新颖类似物的合成。一种新化合物(1)表现出与参比化合物相似的效价,证实了与萘环系统稠合的噻吩并二酮是该分子适应IN活性位点的最佳组合。因此,用氧代替噻唑环中的硫似乎不会显着影响效力。另一方面,在多环系统的位置1处引入额外的甲基或从硫氮平向奥氮平骨架的转移降低了效力。为了了解它们与IN活性位点的相互作用方式,我们将所有化合物对接到先前报道的IN的X射线晶体结构上。我们观察到化合物7-9占据了一个靠近D64和Mg(2+)的区域,并被氨基酸残基K159,K156,N155
-
Derivatives of 2-(iminomethyl)amino-phenyl, their preparation, their use as medicaments and the pharmaceutical compositions containing them申请人:Societe de Conseils de Recherches et d'Applications Scientifiques (S.C.R.A.S.)公开号:US06335445B1公开(公告)日:2002-01-01A compound selected from the group consisting of a compound of the formula wherein A is selected from the group consisting of and the other substituents are defined in the specification having an inhibitory activity of NO-synthase enzymes producing nitrogen mono-oxide and/or an activity which traps the reactive oxygen species.
-
Discovery of ORN0829, a potent dual orexin 1/2 receptor antagonist for the treatment of insomnia作者:Aya Futamura、Ryo Suzuki、Yunoshin Tamura、Hiroshi Kawamoto、Mari Ohmichi、Noriko Hino、Yuichi Tokumaru、Sora Kirinuki、Tetsuaki Hiyoshi、Takeshi Aoki、Daiji Kambe、Dai NozawaDOI:10.1016/j.bmc.2020.115489日期:2020.7design, synthesis, and SAR of dual orexin 1 and 2 receptor antagonists, which were optimized by balancing the antagonistic activity for orexin receptors and lipophilicity. Based on the prototype compound 1, ring construction and the insertion of an additional heteroatom into the resulting ring led to the discovery of orexin 1 and 2 receptor antagonists, which were 3-benzoyl-1,3-oxazinane derivatives. Within在这里,我们介绍了双orexin 1和2受体拮抗剂的设计,合成和SAR,这些受体拮抗剂通过平衡对orexin受体的拮抗活性和亲脂性进行了优化。基于原型化合物1,环的构建以及在所得环中插入一个额外的杂原子导致发现orexin 1和2受体拮抗剂,它们是3-苯甲酰基-1,3-恶嗪烷衍生物。在这些衍生物中,(-)- 3h具有很高的双重orexin受体拮抗活性和低亲脂性。在大鼠多导睡眠图研究中,化合物(-)- 3h在po剂量为1 mg / kg时表现出有效的促睡眠作用,并具有最佳的PK特性和快速的T max观察到大鼠和狗的半衰期短,表明人的半衰期预计为0.9-2.0 h。因此,(-)- 3h(ORN0829;研究代号TS-142)被选为可行的候选药物,目前正处于临床研究中,以治疗失眠症。
-
Synthesis and antihypertensive activity of N-(mercaptoacyl)-thiazolidinecarboxylic acids.作者:MASAYUKI OYA、TOSHIO BABA、EISHIN KATO、YOICHI KAWASHIMA、TOSHIO WATANABEDOI:10.1248/cpb.30.440日期:——The synthesis and antihypertensive activity of a new series of N-(mercaptoacyl)-thiazolidinecarboxylic acids (VIIa-d) are described. Antihypertensive activity was evaluated in terms of angiotensin I-converting enzyme (ACE) inhibitory activity. The activities of these compounds were compared with that of (2S)-1-[(2S)-3-mercapto-2-methylpropanoyl] proline, SQ 14225, and many of them were found to be relatively potent inhibitors of ACE. The most potent was (4R)-2-(2-hydroxyphenyl)-3-(3-mercaptopropanoyl)-4-thiazolidinecarboxylic acid (62). Structure-activity relationships among the thiazolidines and some related compounds are discussed.
-
Bicyclic 6-alkylidene-penems as class-D beta-lactamases inhibitors申请人:Mansour Suhayl Tarek公开号:US20060276445A1公开(公告)日:2006-12-07This invention relates to certain bicyclic 6-alkylidene penems which act as a inhibitor of class-D enzymes. β-Lactamases hydrolyze β-lactam antibiotics, and as such serve as the primary cause of bacterial resistance. The compounds of the present invention when combined with β-lactam antibiotics will provide an effective treatment against life threatening bacterial infections. In accordance with the present invention there are provided compounds of general formula I or a pharmaceutically acceptable salt or in vivo hydrolyzable ester R 5 thereof: wherein: One of A and B denotes hydrogen and the other an optionally substituted fused bicyclic heteroaryl group; and X═O or S.
表征谱图
-
氢谱1HNMR
-
质谱MS
-
碳谱13CNMR
-
红外IR
-
拉曼Raman
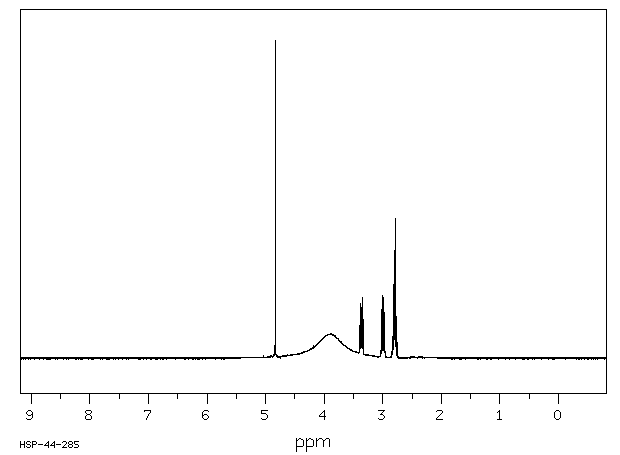
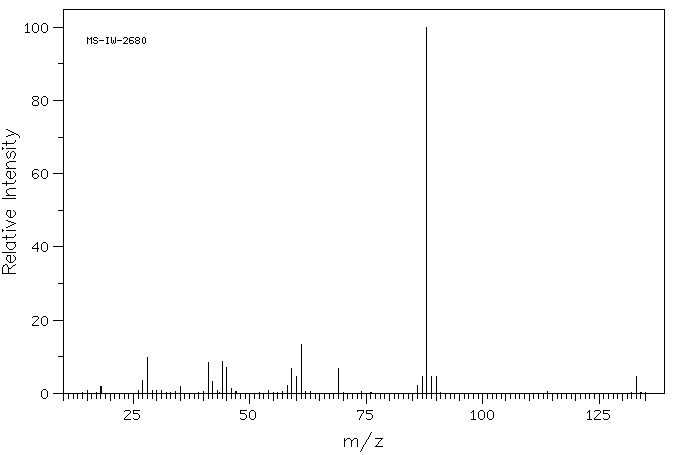
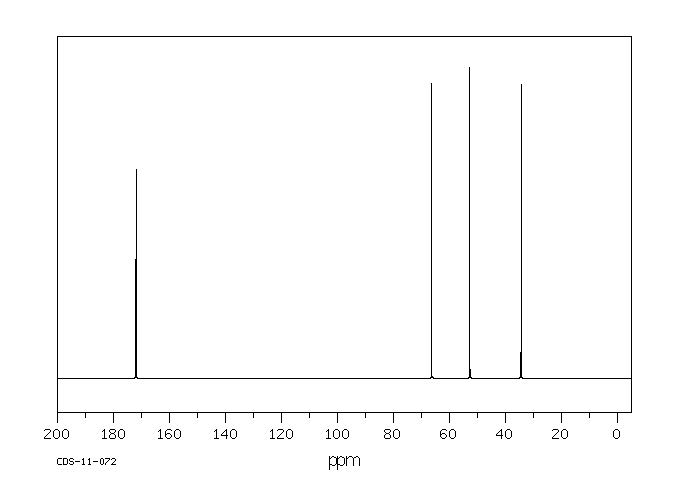
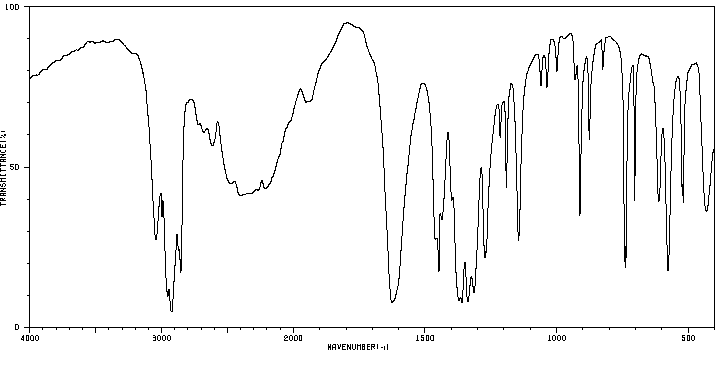
-
峰位数据
-
峰位匹配
-
表征信息
同类化合物
(甲基3-(二甲基氨基)-2-苯基-2H-azirene-2-羧酸乙酯)
(±)-盐酸氯吡格雷
(±)-丙酰肉碱氯化物
(d(CH2)51,Tyr(Me)2,Arg8)-血管加压素
(S)-(+)-α-氨基-4-羧基-2-甲基苯乙酸
(S)-阿拉考特盐酸盐
(S)-赖诺普利-d5钠
(S)-2-氨基-5-氧代己酸,氢溴酸盐
(S)-2-[[[(1R,2R)-2-[[[3,5-双(叔丁基)-2-羟基苯基]亚甲基]氨基]环己基]硫脲基]-N-苄基-N,3,3-三甲基丁酰胺
(S)-2-[3-[(1R,2R)-2-(二丙基氨基)环己基]硫脲基]-N-异丙基-3,3-二甲基丁酰胺
(S)-1-(4-氨基氧基乙酰胺基苄基)乙二胺四乙酸
(S)-1-[N-[3-苯基-1-[(苯基甲氧基)羰基]丙基]-L-丙氨酰基]-L-脯氨酸
(R)-乙基N-甲酰基-N-(1-苯乙基)甘氨酸
(R)-丙酰肉碱-d3氯化物
(R)-4-N-Cbz-哌嗪-2-甲酸甲酯
(R)-3-氨基-2-苄基丙酸盐酸盐
(R)-1-(3-溴-2-甲基-1-氧丙基)-L-脯氨酸
(N-[(苄氧基)羰基]丙氨酰-N〜5〜-(diaminomethylidene)鸟氨酸)
(6-氯-2-吲哚基甲基)乙酰氨基丙二酸二乙酯
(4R)-N-亚硝基噻唑烷-4-羧酸
(3R)-1-噻-4-氮杂螺[4.4]壬烷-3-羧酸
(3-硝基-1H-1,2,4-三唑-1-基)乙酸乙酯
(2S,4R)-Boc-4-环己基-吡咯烷-2-羧酸
(2S,3S,5S)-2-氨基-3-羟基-1,6-二苯己烷-5-N-氨基甲酰基-L-缬氨酸
(2S,3S)-3-((S)-1-((1-(4-氟苯基)-1H-1,2,3-三唑-4-基)-甲基氨基)-1-氧-3-(噻唑-4-基)丙-2-基氨基甲酰基)-环氧乙烷-2-羧酸
(2S)-2,6-二氨基-N-[4-(5-氟-1,3-苯并噻唑-2-基)-2-甲基苯基]己酰胺二盐酸盐
(2S)-2-氨基-N,3,3-三甲基-N-(苯甲基)丁酰胺
(2S)-2-氨基-3-甲基-N-2-吡啶基丁酰胺
(2S)-2-氨基-3,3-二甲基-N-(苯基甲基)丁酰胺,
(2S)-2-氨基-3,3-二甲基-N-2-吡啶基丁酰胺
(2S,4R)-1-((S)-2-氨基-3,3-二甲基丁酰基)-4-羟基-N-(4-(4-甲基噻唑-5-基)苄基)吡咯烷-2-甲酰胺盐酸盐
(2R,3'S)苯那普利叔丁基酯d5
(2R)-2-氨基-3,3-二甲基-N-(苯甲基)丁酰胺
(2-氯丙烯基)草酰氯
(1S,3S,5S)-2-Boc-2-氮杂双环[3.1.0]己烷-3-羧酸
(1R,5R,6R)-5-(1-乙基丙氧基)-7-氧杂双环[4.1.0]庚-3-烯-3-羧酸乙基酯
(1R,4R,5S,6R)-4-氨基-2-氧杂双环[3.1.0]己烷-4,6-二羧酸
齐特巴坦
齐德巴坦钠盐
齐墩果-12-烯-28-酸,2,3-二羟基-,苯基甲基酯,(2a,3a)-
齐墩果-12-烯-28-酸,2,3-二羟基-,羧基甲基酯,(2a,3b)-(9CI)
黄酮-8-乙酸二甲氨基乙基酯
黄荧菌素
黄体生成激素释放激素(1-6)
黄体生成激素释放激素 (1-5) 酰肼
黄体瑞林
麦醇溶蛋白
麦角硫因
麦芽聚糖六乙酸酯
麦根酸
相关功能分类

联系我们



关注我们

公众号
