Citalopram is metabolized mainly in the liver via <i>N</i>-demethylation to its main metabolite, _demethylcitalopram_ by CYP2C19 and CYP3A4. Other metabolites include _didemethylcitalopram_ via CYP2D6 metabolism, and _citalopram <i>N</i>-oxide_ via monoamine oxidase enzymes and aldehyde oxidase. It is a deaminated propionic acid derivative. After a single dose of citalopram, peak blood concentrations occur at approximately 4 hours. This drug in is found mainly unchanged in the plasma as citalopram. Cytochrome P450 (CYP) 3A4 and 2C19 isozymes appear to be heavily involved in producing _demethylcitalopram_. Demethylcitalopram appears to be further <i>N</i>-demethylated by CYP2D6 to didemethylcitalopram. Citalopram metabolites exert little pharmacologic activity in comparison to the parent drug and are not likely to contribute to the clinical effect of citalopram.
In a 2002 study, 11 women took citalopram (20-40 mg/day) during pregnancy; 10 throughtout gestation and 1 starting at 20 weeks' gestation. The mean ratio of two metabolites was significantly higher during pregnancy than at 2 months postdelivery, indicating induction of the CYP2D6 isoenzyme. The trough plasma concentration of citalopram, desmethylcitalopram, and didesmethylcitalopram in the normal new borns were 64%, 66%, and 68% of the maternal concentrations, respectively.
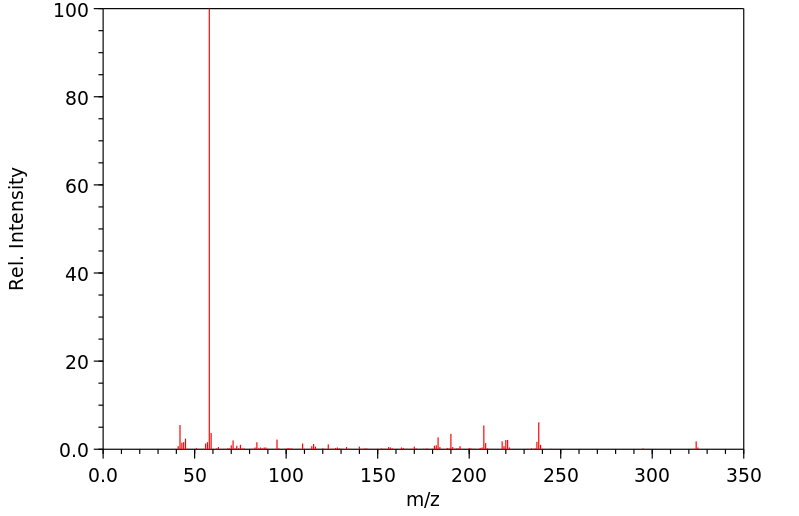
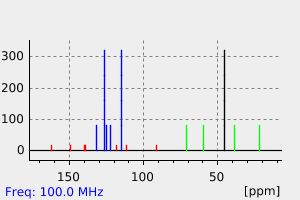
The antidepressant citalopram (CT), a selective serotonin uptake inhibitor, was given in its labelled form, 14(C)-CT, as a single oral dose in 50 mL aqueous solution (0.1 mmol/30 uCi/1.1 MBq) to four healthy male volunteers. Concentrations of radioactivity in whole blood and plasma were similar. ... The HPLC profile of urinary components showed that besides the known metabolites of citalopram, three glucuronides were present. The relative amounts of CT and its metabolites in urine collected for 7 days were: CT (26 %), N-demethyl-CT (DCT, 19%), N,N-didemethyl-CT (DDCT,9%), the N-oxide (7%), the quaternary ammonium glucuronide of CT (CT-GLN, 14%), the N-glucuronide of DDCT (DDCT-GLN, 6%), and the glucuronide of the acid metabolite (CT-acid-GLN, 12%) formed by N,N-dimethyl deamination of CT. CT-GLN was isolated using preparative chromatography and identified by LC-MS-MS and NMR. DDCT-GLN and CT-acid-GLN were identified by LC-MS. This study shows that protracted renal excretion represents the major route of elimination, with a small fraction voided with feces. A considerable portion of the urinary excreted dose consists of N-glucuronides of CT and DDCT together with the O-acyl glucuronide of CT-acid.
This study was conducted to identify enzyme systems eventually catalyzing a local cerebral metab of citalopram. ... The metab of citalopram, of its enantiomers and demethylated metabolites was investigated in rat brain microsomes & in rat and human brain mitochondria. No cytochrome P-450 mediated transformation was observed in rat brain. ... In rat whole brain and in human frontal cortex, putamen, cerebellum & white matter of five brains monoamine oxidase activity was determined by the stereoselective measurement of the production of citalopram propionate. All substrates were metabolized by both forms of MAO, except in rat brain, where monoamine oxidase B activity could not be detected. Apparent Km and Vmax of S-citalopram biotransformation in human frontal cortex by monoamine oxidase B were found to be 266 uM & 6.0 pmol min/mg protein & by monoamine oxidase A 856 uM & 6.4 pmol min/mg protein, respectively. These Km values are in the same range as those for serotonin & dopamine metab by monoamine oxidases. ...
Citalopram ... is N-demethylated to N-desmethylcitalopram partially by CYP2C19 & partially by CYP3A4 & N-desmethylcitalopram is further N-demethylated by CYP2D6 to the likewise inactive metabolite di-desmethylcitalopram. The two metabolites are not active. ... In vitro citalopram does not inhibit CYP or does so only very moderately. A number of studies in healthy subjects and patients have confirmed, that this also holds true in vivo. Thus no change in pharmacokinetics or only very small changes were observed when citalopram was given with CYP1A2 substrates (clozapine & therophylline), CYP2C9 (warfarin), CYP2C19 (imipramine & mephenytoin), CYP2D6 (sparteine, imipramine & amitriptyline) and CYP3A4 (carbamazepine & triazolam). ...
IDENTIFICATION AND USE: Citalopram is a solid. It is a serotonin uptake inhibitor, and second-generation antidepressive agent. Citalopram tablets are indicated for the treatment of depression. HUMAN STUDIES: Symptoms most often accompanying citalopram overdose, alone or in combination with other drugs and/or alcohol, included dizziness, sweating, nausea, vomiting, tremor, somnolence, and sinus tachycardia. In more rare cases, observed symptoms included amnesia, confusion, coma, convulsions, hyperventilation, cyanosis, rhabdomyolysis, and ECG changes (including QTc prolongation, nodal rhythm, ventricular arrhythmia, and very rare cases of torsade de pointes). Acute renal failure has been very rarely reported accompanying overdose. Five male infants exposed to citalopram (30 mg/day), paroxetine (10-40 mg/day) or fluoxetine (20 mg/day) during gestation exhibited withdrawal symptoms at or within a few days of birth and lasting up to 1 month. Symptoms included irritability, constant crying, shivering, increased tonus, eating and sleeping problems, and convulsions. Late gestational exposure to citalopram, may be associated with a neonatal toxicity syndrome with immediate onset at birth or soon after birth and sometimes may be mistaken for neonatal withdrawal syndrome. A 3860 g infant was delivered at 40 weeks gestation. The mother had been taking citalopram 20 mg/day until the day of delivery. Citalopram was not clastogenic in the in vitro chromosomal aberration assay in human lymphocytes. ANIMAL STUDIES: Citalopram was administered in the diet to mice and rats for 18 and 24 months, respectively. There was no evidence for carcinogenicity of citalopram in mice receiving up to 240 mg/kg/day. There was an increased incidence of small intestine carcinoma in rats receiving 8 or 24 mg/kg/day doses. The relevance of these findings to humans is unknown. Pathologic changes (degeneration/atrophy) were observed in the retinas of albino rats in the 2-year carcinogenicity study with citalopram. In a rabbit study, no adverse effects on embryo/fetal development were observed at doses of up to 16 mg/kg/day. When female rats were treated with citalopram (4.8, 12.8, or 32 mg/kg/day) from late gestation through weaning, increased offspring mortality during the first 4 days after birth and persistent offspring growth retardation were observed at the highest dose. In two rat embryo/fetal development studies, oral administration of citalopram (32, 56, or 112 mg/kg/day) to pregnant animals during the period of organogenesis resulted in decreased embryo/fetal growth and survival and an increased incidence of fetal abnormalities (including cardiovascular and skeletal defects) at the high dose. Citalopram was mutagenic in the in vitro bacterial reverse mutation assay (Ames test) in 2 of 5 bacterial strains (Salmonella TA98 and TA1537) in the absence of metabolic activation. It was clastogenic in the in vitro Chinese hamster lung cell assay for chromosomal aberrations in the presence and absence of metabolic activation. Citalopram was not mutagenic in the in vitro mammalian forward gene mutation assay (HPRT) in mouse lymphoma cells or in a coupled in vitro/in vivo unscheduled DNA synthesis (UDS) assay in rat liver.
The antidepressant, antiobsessive-compulsive, and antibulimic actions of citalopram are presumed to be linked to its inhibition of CNS neuronal uptake of serotonin. Citalopram blocks the reuptake of serotonin at the serotonin reuptake pump of the neuronal membrane, enhancing the actions of serotonin on 5HT<sub>1A</sub> autoreceptors. SSRIs bind with significantly less affinity to histamine, acetylcholine, and norepinephrine receptors than tricyclic antidepressant drugs.
Rapidly and well absorbed from the GI tract. Peak plasma concentrations occur within 4 hours of a single orally administered dose. Bioavailability is 80% following oral administration. Food does not affect absorption.
12 L/kg Citalopram is highly lipophilic and likely widely distributed throughout the body, including the blood-brain-barrier. However, its metabolite, _demethylcitalopram_ does not penetrate the blood-brain-barrier well.
Like other selective serotonin-reuptake inhibitors, citalopram is a highly lipophilic compound that appears to be rapidly and well absorbed from the GI tract following oral administration. Following a single 40-mg oral dose of citalopram as a tablet, the manufacturer states that peak plasma concentrations averaging approximately 44 ng/mL occur at about 4 hours.
Structure−Activity Relationships for a Novel Series of Citalopram (1-(3-(Dimethylamino)propyl)-1-(4-fluorophenyl)-1,3-dihydroisobenzofuran-5-carbonitrile) Analogues at Monoamine Transporters
[EN] SUBSTITUTED N-HETEROCYCLIC CARBOXAMIDES AS ACID CERAMIDASE INHIBITORS AND THEIR USE AS MEDICAMENTS<br/>[FR] CARBOXAMIDES N-HÉTÉROCYCLIQUES SUBSTITUÉS UTILISÉS EN TANT QU'INHIBITEURS DE LA CÉRAMIDASE ACIDE ET LEUR UTILISATION EN TANT QUE MÉDICAMENTS
申请人:BIAL BIOTECH INVEST INC
公开号:WO2021055627A1
公开(公告)日:2021-03-25
The invention provides substituted N-heterocyclic carboxamides and related compounds, compositions containing such compounds, medical kits, and methods for using such compounds and compositions to treat a medical disorder, e.g., cancer, lysosomal storage disorder, neurodegenerative disorder, inflammatory disorder, in a patient.
[EN] COMPOUNDS AND THEIR USE AS BACE INHIBITORS<br/>[FR] COMPOSÉS ET LEUR UTILISATION EN TANT QU'INHIBITEURS DE BACE
申请人:ASTRAZENECA AB
公开号:WO2016055858A1
公开(公告)日:2016-04-14
The present application relates to compounds of formula (I), (la), or (lb) and their pharmaceutical compositions/preparations. This application further relates to methods of treating or preventing Αβ-related pathologies such as Down's syndrome, β- amyloid angiopathy such as but not limited to cerebral amyloid angiopathy or hereditary cerebral hemorrhage, disorders associated with cognitive impairment such as but not limited to MCI ("mild cognitive impairment"), Alzheimer's disease, memory loss, attention deficit symptoms associated with Alzheimer's disease, neurodegeneration associated with diseases such as Alzheimer's disease or dementia, including dementia of mixed vascular and degenerative origin, pre-senile dementia, senile dementia and dementia associated with Parkinson's disease.
[EN] METHYL OXAZOLE OREXIN RECEPTOR ANTAGONISTS<br/>[FR] MÉTHYLOXAZOLES ANTAGONISTES DU RÉCEPTEUR DE L'OREXINE
申请人:MERCK SHARP & DOHME
公开号:WO2016089721A1
公开(公告)日:2016-06-09
The present invention is directed to methyl oxazole compounds which are antagonists of orexin receptors. The present invention is also directed to uses of the compounds described herein in the potential treatment or prevention of neurological and psychiatric disorders and diseases in which orexin receptors are involved. The present invention is also directed to compositions comprising these compounds. The present invention is also directed to uses of these compositions in the potential prevention or treatment of such diseases in which orexin receptors are involved.
NAPHTHALENE-BASED INHIBITORS OF ANTI-APOPTOTIC PROTEINS
申请人:Pellecchia Maurizio
公开号:US20090105319A1
公开(公告)日:2009-04-23
Methods of using apogossypol and its derivatives for treating inflammation is disclosed. Also, there is described a group of compounds having structure A, or a pharmaceutically acceptable salt, hydrate, N-oxide, or solvate thereof are provided:
wherein each R is independently selected from the group consisting of H, C(O)X, C(O)NHX, NH(CO)X, SO
2
NHX, and NHSO
2
X, wherein X is selected from the group consisting of an alkyl, a substituted alkyl, an aryl, a substituted aryl, an alkylaryl, and a heterocycle. Compounds of group A may be used for treating various diseases or disorders, such as cancer.