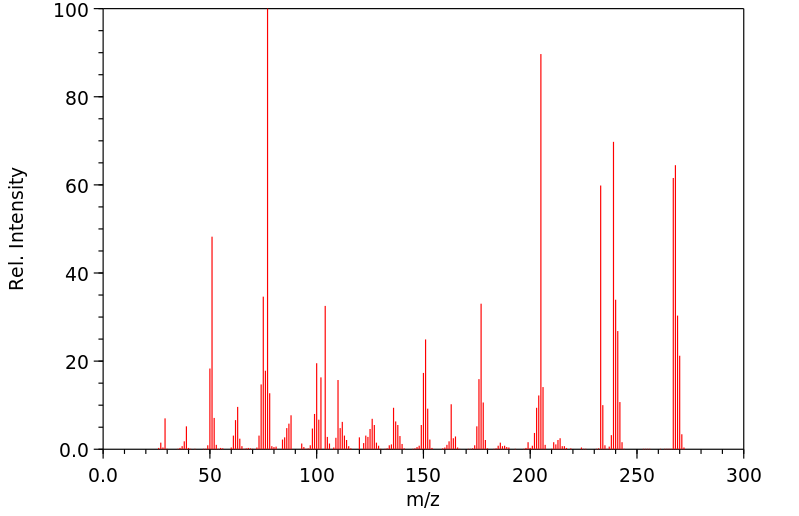
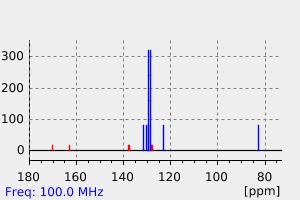
Oxazepam has a single major inactive metabolite, a glucuronide conjugate. The glucuronidation of the S-isomer is catalyzed by UGT2B15. The glucuronidation of the R-isomer is catalyzed by UGT2B7 and UGT1A9.
The metabolism and the anticonvulsant effect of clorazepate were followed for 2 h after its i.v. administration to mice. The ED50 of the drug was 12 mg/kg at 1 min against pentetrazole-induced convulsions (45 mg/kg i.v.), it reached a minimum at 1 hr (2.0 mg/kg) and rose to 2.7 mg/kg at 2 h. The concentrations of unchanged clorazepate and its metabolites, desmethyldiazepam and oxazepam, were determined in plasma and brain after administration of the respective ED50s. Unchanged clorazepate could be detected in plasma for the first hour but never in brain, so it can be considered as inactive pro-drug. The brain concentrations of desmethyldiazepam and oxazepam after the respective ED50s of clorazepate were considerably higher at 1 and 15 min than after longer time intervals. This may be explained by a time lag needed to reach and bind to the benzodiazepine receptor.
Oxazepam is a commonly used 1,4-benzodiazepine anxiolytic drug that is polymorphically metabolized in humans. However, the molecular basis for this phenomenon is currently unknown. We have previously shown that S-oxazepam glucuronide, the major oxazepam metabolite, is selectively formed by UDP-glucuronosyltransferase (UGT) 2B15, whereas the minor Roxazepam glucuronide is produced by multiple UGTs other than UGT2B15. Phenotype-genotype studies were conducted using microsomes and DNA prepared from the same set of 54 human livers. Sequencing of the UGT2B15 gene revealed three nonsynonymous polymorphisms, D85Y, T352I, and K523T, with variant allele frequencies of 0.56, 0.02, and 0.40, respectively. D85Y genotype showed a significant effect (p = 0.012) on S-oxazepam glucuronidation with lower median activities in 85Y/Y livers (49 pmol/min/mg protein) compared with 85D/D livers (131 pmol/min/mg), whereas 85D/Y livers were intermediate in activity (65 pmol/min/mg). There was also a significant trend (p = 0.049) for higher S-oxazepam activities in the two 352T/I livers (135 and 210 pmol/min/mg) compared with the remaining 352T/T livers (median, 64 pmol/min/mg). Conversely, K523T genotype had no apparent effect on oxazepam glucuronidation (p > 0.05). Donor gender also significantly influenced S-oxazepam glucuronidation with higher median activities in male (65 pmol/min/mg) compared with female (39 pmol/min/ mg) livers (p = 0.042). R-Oxazepam glucuronidation was not affected by either genotype or gender (p > 0.05). In conclusion, gender and D85Y genotype are identified as major determinants of S-oxazepam glucuronidation by human liver and may explain in part polymorphic oxazepam glucuronidation by human subjects.
There are three major pathways of oxazepam metabolism in mice and rats (as in humans): direct conjugation, phenyl ring oxidation and diazepine ring contraction. In mice, conjugation is mainly with glucuronide, predominantly excreted in the urine; in rats, conjugation is mainly with sulfate, which is almost entirely eliminated in the feces. The sulfate conjugate of oxazepam, which is unstable in acidic media, may be the source of the fecal oxazepam It has not been detected in mice. Studies with recirculating, perfused male Swiss (CD-1) mouse liver preparations showed that oxazepam glucuronides are the dominant liver metabolites in this species. Oxazepam can also be conjugated with glucuronide by the placenta of rabbits, apparently in contrast to the human organ. Phenyl ring oxidation is more important in rats than in mice (or humans) and a dihydrodiol (probably the 3',4'- dihydrodiol, since 2'-hydroxy derivatives are not known) accounts for about 30% of the 72-hr urinary metabolites in Fischer 344 rats. This metabolite, which probably forms via an arene oxide intermediate and has not been found in mice, holds implications for the toxicological properties of oxazepam. In rats, ring contraction to 6-chloro-4-phenyl- 2(1H)-quinazoline carboxylic acid occurs to roughly one half of the extent seen in mice.
Similar to other benzodiazepines, oxazepam exerts its anxiolytic effects by potentiating the effect of gamma-aminobutyric acid (GABA) on GABA-A receptors through a cooperative mechanism of action. GABA receptors are ionotropic chloride-linked channel receptors that produce inhibitory postsynaptic potentials. When activated by GABA, the GABA receptor/chloride ionophore complex undergoes a conformational change that allows the passage of chloride ions through the channel. Benzodiazepines are believed to exert their effect by increasing the effect of GABA at its receptor. Benzodiazepine binding increases chloride conductance in the presence of GABA by increasing the frequency at which the channel opens. In contrast, barbiturates increase chloride conductance in the presence of GABA by prolonging the time in which the channel remains open. There are 18 subtypes of the GABA receptor subunits. The α<sub>2</sub> subunit of the α<sub>2</sub>β<sub>3</sub>γ<sub>2</sub> receptor complex is thought to mediate anxiolytic effects while the α<sub>1</sub> subunit of the α<sub>1</sub>β<sub>2</sub>γ<sub>2</sub> receptor complex is thought to mediate sedative, anticonvulsant and anterograde amnesia effects.
Oxazepam, like other benzodiazepines, is rarely associated with serum ALT or alkaline phosphatase elevations, and clinically apparent liver injury from oxazepam is extremely rare, if it occurs at all. Despite its availability for more than 50 years, there have been no case reports of symptomatic, acute liver injury from oxazepam. Nevertheless, the possibility of liver dysfunction and jaundice are mentioned in the product label for oxazepam. Isolated single cases of clinically apparent liver injury have been reported with other benzodiazepines including alprazolam, chlordiazepoxide, clonazepam, diazepam, flurazepam, lorazepam, and triazolam. The clinical pattern of acute liver injury from benzodiazepines is typically cholestatic and mild-to-moderate in severity with a latency of 1 to 6 months. Fever and rash are uncommon as is autoantibody formation.
Oxazepam is primarily eliminated in the urine as its glucuronide metabolite, with the feces containing approximately 21% of the unchanged drug. The majority of an orally ingested dose of oxazepam is excreted within 48 hours.
The miniature swine (like humans) eliminated oxazepam primarily as the glucuronides, while aromatic hydroxylation predominated in the rat. In rats, 70.7 +/- 6.0% of a single oral dose of 20 mg/kg bw was eliminated in feces following biliary secretion, while 18.9 +/- 2.4% of the dose was found in the urine. In CD-1 mice given an oral dose of 22 mg/kg bw oxazepam, 57.8% was recovered from the feces and 27.3% was recovered from urine over five days. ... Treatment with 2500 mg/kg diet (ppm) oxazepam in the diet for 14 days before administration of oxazepam by gastric instillation led to a shift from fecal to urinary excretion in mice, but not rats, so that the urinary excretion almost doubled.
Oxazepam accumulates in adipose tissue. /It was/ found that adipose tissue/blood ratios of the drug in mice given 5 mg/kg bw intravenously varied from 1.7 (at 5 min) to 4.9 (at 30 min). Accumulation also occurred in the brain. Maximal concentrations of oxazepam in the brain were 14.3 +/- 0.17 ug/g in mice, 4.5 +/- 0.03 ug/g in rats and 3.5 +/- 0.47 ug/g in guinea-pigs, all at 5 min. Brain/blood drug level ratios in these species varied from 1.1 (at 1 min) to 11.3 (at 10 hr) in mice, from 1.9 (at 1 min) to 6.2 (at 1 hr) in rats and from 1.9 (at 5 min) to 8.9 (at 5 hr) in guinea-pigs.
/It was/ found that the time of maximum absorption of 30 mg oxazepam was 2.2 hr (range, 0.75-4.25 hr) in 18 men and 3.1 hr (range, 0.5-8.0 hr) in 20 women. The maximal plasma concentrations in this study were 622 +/- 37 ng/mL in men and 837 +/- 51 ng/mL in women.
[EN] COMPOUNDS AND THEIR USE AS BACE INHIBITORS<br/>[FR] COMPOSÉS ET LEUR UTILISATION EN TANT QU'INHIBITEURS DE BACE
申请人:ASTRAZENECA AB
公开号:WO2016055858A1
公开(公告)日:2016-04-14
The present application relates to compounds of formula (I), (la), or (lb) and their pharmaceutical compositions/preparations. This application further relates to methods of treating or preventing Αβ-related pathologies such as Down's syndrome, β- amyloid angiopathy such as but not limited to cerebral amyloid angiopathy or hereditary cerebral hemorrhage, disorders associated with cognitive impairment such as but not limited to MCI ("mild cognitive impairment"), Alzheimer's disease, memory loss, attention deficit symptoms associated with Alzheimer's disease, neurodegeneration associated with diseases such as Alzheimer's disease or dementia, including dementia of mixed vascular and degenerative origin, pre-senile dementia, senile dementia and dementia associated with Parkinson's disease.
[EN] METHYL OXAZOLE OREXIN RECEPTOR ANTAGONISTS<br/>[FR] MÉTHYLOXAZOLES ANTAGONISTES DU RÉCEPTEUR DE L'OREXINE
申请人:MERCK SHARP & DOHME
公开号:WO2016089721A1
公开(公告)日:2016-06-09
The present invention is directed to methyl oxazole compounds which are antagonists of orexin receptors. The present invention is also directed to uses of the compounds described herein in the potential treatment or prevention of neurological and psychiatric disorders and diseases in which orexin receptors are involved. The present invention is also directed to compositions comprising these compounds. The present invention is also directed to uses of these compositions in the potential prevention or treatment of such diseases in which orexin receptors are involved.
Heterobicyclic compounds of Formula (I):
or a pharmaceutically-acceptable salt, tautomer, or stereoisomer thereof, as defined in the specification, and compositions containing them, and processes for preparing such compounds. Provided herein also are methods of treating disorders or diseases treatable by inhibition of PDE10, such as obesity, non-insulin dependent diabetes, schizophrenia, bipolar disorder, obsessive-compulsive disorder, Huntington's Disease, and the like.
Formula (I)的杂环化合物:
或其药用可接受的盐、互变异构体或立体异构体,如规范中所定义,并含有它们的组合物,以及制备这种化合物的方法。本文还提供了通过抑制PDE10来治疗由此可治疗的疾病或疾病的方法,如肥胖症、非胰岛素依赖型糖尿病、精神分裂症、躁郁症、强迫症、亨廷顿病等。
[EN] IMIDAZOLIUM REAGENT FOR MASS SPECTROMETRY<br/>[FR] RÉACTIF D'IMIDAZOLIUM POUR SPECTROMÉTRIE DE MASSE
申请人:HOFFMANN LA ROCHE
公开号:WO2021234004A1
公开(公告)日:2021-11-25
The present invention relates to compounds which are suitable to be used in mass spectrometry as well as methods of mass spectrometric determination of analyte molecules using said compounds.
本发明涉及适用于质谱的化合物,以及利用该化合物进行分析物分子的质谱测定方法。
[EN] NAPHTHALENE CARBOXAMIDE M1 RECEPTOR POSITIVE ALLOSTERIC MODULATORS<br/>[FR] COMPOSÉS DE NAPHTHALÈNE CARBOXAMIDE, MODULATEURS ALLOSTÉRIQUES POSITIFS DU RÉCEPTEUR M1
申请人:MERCK SHARP & DOHME
公开号:WO2011149801A1
公开(公告)日:2011-12-01
The present invention is directed to naphthalene carboxamide compounds of formula (I) which are M1 receptor positive allosteric modulators and that are useful in the treatment of diseases in which the M1 receptor is involved, such as Alzheimers disease, schizophrenia, pain or sleep disorders. The invention is also directed to pharmaceutical compositions comprising the compounds and to the use of the compounds and compositions in the treatment of diseases mediated by the M1 receptor.