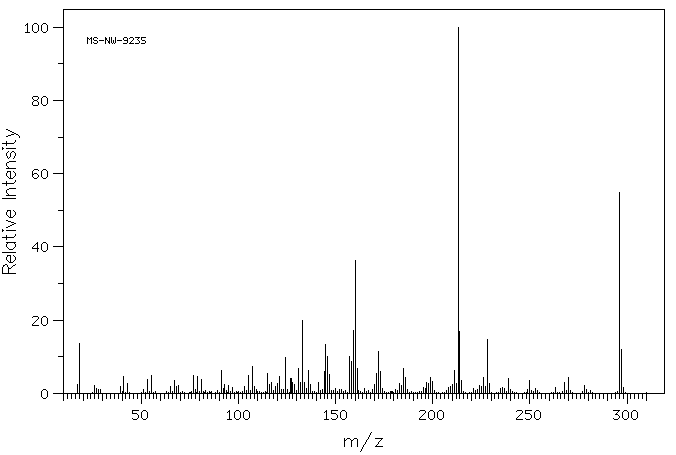
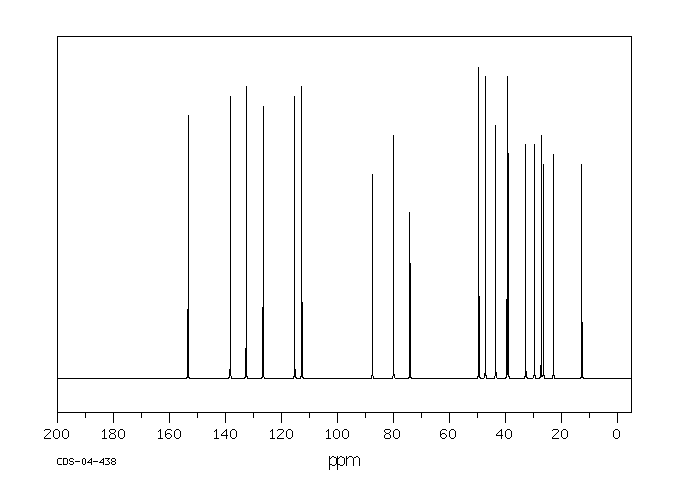
Ethinylestradiol can be glucuronidated by UGT1A1, UGT1A3, UGT1A4, UGT1A9, and UGT2B7. Ethinylestradiol is also sulfated by SULT1A1, SULT1A3, and SULT1E1. Ethinylestradiol can also be hydroxylated at positions 2, 4, 6, 7, and 16 by CYP3A4, CYP3A5, CYP2C8, CYP2C9, and CYP1A2. These hydroxylated metabolites can be methylated by catechol-O-methyltransferase. The methoxy metabolites can in turn be sulfated or glucuronidated.
Exogenous estrogens are metabolized in the same manner as endogenous estrogens. Circulating estrogens exist in a dynamic equilibrium of metabolic interconversions. These transformations take place mainly in the liver. Estradiol is converted reversibly to estrone, and both can be converted to estriol, which is the major urinary metabolite. Estrogens also undergo enterohepatic recirculation via sulfate and glucuronide conjugation in the liver, biliary secretion of conjugates into the intestine, and hydrolysis in the gut followed by reabsorption. In postmenopausal women, a significant proportion of the circulating estrogens exist as sulfate conjugates, especially estrone sulfate, which serves as a circulating reservoir for the formation of more active estrogens.
Ethinyl estradiol is extensively metabolized, both by oxidation and by conjugation with sulfate and glucuronide. Sulfates are the major circulating conjugates of ethinyl estradiol and glucuronides predominate in urine. The primary oxidative metabolite is 2-hydroxy ethinyl estradiol, formed by the CYP3A4 isoform of cytochrome P450. Part of the first-pass metabolism of ethinyl estradiol is believed to occur in gastrointestinal mucosa. Ethinyl estradiol may undergo enterohepatic circulation.
Studies on the metabolism of ethinylestradiol have been carried out in rats, rabbits, guinea-pigs, dogs and monkeys. It is very rapidly and effectively absorbed from rat intestine; no appreciable metabolic transformation is reported to take place during the absorption process. The main metabolic pathway of ethinylestradiol in rats is by aromatic 2-hydroxylation; hydroxylations at ring B (C-6/C-7) are of only minor importance. Rat liver forms 2-hydroxyethinylestradiol and the methyl ethers thereof, 2-methoxyethinylestradiol and 2-hydroxyethinylestradiol-3-methy1 ether, as its major metabolic products. This pathway is also important in humans. Metabolites of ethinylestradiol in rats are excreted almost exclusively in the feces.
IDENTIFICATION: Ethinylestradiol is a synthetic steroid, prepared from estrone. It is a white to creamy or slightly yellowish-white powder or crystals. It is insoluble in water, soluble in ethanol. Indications: The most frequent use is as the estrogen component of combined oral contraceptives. Also used for the treatment of menopausal and post menopausal symptoms, especially the vasomotor effects. Used in the treatment of female hypogonadism and as a palliative treatment in malignant neoplasm of breast and prostate. Also used in the treatment of some women with acne, and in Turner's syndrome. HUMAN EXPOSURE: Main risks and target organs: Acute poisoning with ethinylestradiol results in mild, self limiting effects, usually involving the gastrointestinal tract. Chronic toxicity increases the risk of cardiovascular disease, including myocardial infarction, cerebrovascular disease, thromboembolic disease, gallbladder disease, and certain cancers in some people. Summary of clinical effects: Acute poisoning with ethinylestradiol is mild and self limiting. Nausea, vomiting and occasionally vaginal breakthrough bleeding may occur. Chronic toxicity from ethinylestradiol, like other estrogens, increases the risk for stroke, myocardial infarction and thromboembolic disease in certain populations. Jaundice, hypertension, nasal congestion, headache, dizziness and fluid retention may occur. Endometrial, breast, and certain liver cancers may occur at a higher incidence than the general population. Contraindications: Contraindications are those of the use of estrogens in general, and include the following: Known or suspected carcinoma of the breast, except in selected patients being treated for metastatic disease. Known or suspected estrogen dependent neoplasia. Known or suspected pregnancy. Undiagnosed abnormal genital bleeding. Active thrombophlebitis or thromboembolic disease. A past history of thrombophlebitis, thrombosis or thromboembolic disease associated with the previous use of estrogen containing compounds. Absorption by route of exposure: Ethinylestradiol is rapidly and completely absorbed from the gastrointestinal tract. The ethinyl substitution in the C17 position inhibits first-pass metabolism. Bioavailability is reported at 40%. Distribution by route of exposure: Extensively plasma protein bound, mainly to albumin. Unbound molecules distribute widely in the tissues due to their lipophilic nature. Peak plasma concentrations occur initially at 2 to 3 hours after oral ingestion. A second, 12 hour peak, is thought to represent extensive enterohepatic circulation. Biological half-life by route of exposure: Biological half life is approximately 7.7 hours following a single oral therapeutic dose. Elimination phase half life is reported between 13 and 27 hours. Metabolism: Compared to other estrogens, metabolism is slow. Primary route of biotransformation is via 2-hydroxylation and the formation of 2- and 3-methyl ethers. First-pass metabolism occurs primarily in the gut wall. Elimination by route of exposure: Some enterohepatic circulation of sulfate and glucuronide metabolites does occur, hence some is excreted via the feces. Excretion is also via the kidneys. Mode of action: Toxicodynamics: 2 to 3 fold increase in the incidence of gallbladder disease is reported with the use of estrogens. This is thought due to an increased saturation of bile with cholesterol and a reduction of bile acid secretion. Also, many studies have been performed investigating the adverse effects of estrogens, including ethinylestradiol, on coagulation. These have used estrogens alone, and estrogens in combination with progestins. However a consensus of the net outcome of physiologic or pharmacological doses has not occurred as yet. Pharmacodynamics: Like other steroid hormones, ethinylestradiol is thought to act primarily through the regulation of gene expression. As a lipophilic hormone, it diffuses readily through cellular membranes to bind to estrogen receptors situated in the nucleus. Estrogen receptors are found in the female reproductive tract, breast, pituitary, hypothalamus, bone, liver and other tissues. The receptor interacts with a specialized nucleotide sequence, resulting in either an increase or decrease in the transcription of hormone regulated genes. Tissues may vary in the way in which they respond to receptor activation. Desirable therapeutic effects, include its action on the female reproductive tract, (usually in combination with a progesterone), where ethinylestradiol stimulates proliferation and differentiation in the fallopian tube, and increase the tubal muscular activity. Ethinylestradiol also increases the water content of cervical mucus and favors contraction of the uterine myometrium. Estrogens, including ethinylestradiol, block resorption of bone, resulting in a positive effect on bone mass. It is well established that the risk of endometrial hyperplasia and cancer is increased in women receiving unopposed estrogen replacement therapy, including ethinylestradiol. Data from the 1970's and 1980's reported a 2 to 15 fold increase in the risk of endometrial carcinoma. The higher the dose and the longer the length of therapy the greater the risk. However, the addition of progestogen to estrogen replacement therapy was protective. More research is needed before a conclusion can be drawn on whether ethinylestradiol therapy, and other estrogens, increase the risk of breast carcinoma. Conflicting findings have been reported. A review of data from the 1970's and 1980's suggests that there is a moderate increase in the risk of breast carcinoma, but this did not occur until after 5 years of therapy. Teratogenicity: No specific data available for ethinylestradiol. Reports suggest a link between fetal exposure to female sex hormones and congenital abnormalities. These include heart defects, and limb defects. Other estrogens, namely diethylstilbestrol, have been associated with the development of vaginal and cervical adenocarcinoma in female offspring of mothers who had taken this drug during the first trimester. Diethylstilbestrol ingestion during pregnancy is also associated with a number of other abnormalities in male offspring, including, smaller testes and urogenital abnormalities. Although no studies relating ethinylestradiol directly to these findings were identified, the pharmacological similarities in this class of compounds suggest caution should be used. Main adverse effects: The main adverse effects of ethinylestradiol given in therapeutic dose are directly related to its estrogenic and metabolic effects. They include water and sodium retention, which may result in edema, weight gain and tender breast enlargement. Changes in libido, and withdrawal vaginal bleeding is also reported. Liver function impairment, jaundice and gallstones may occur. Headache, depression, dizziness, glucose intolerance, and a sensitivity to contact lenses are described. Large doses may produce hypercalcemia when used in the treatment of metastatic carcinoma. Nausea, vomiting and diarrhea are not uncommon. Dermatological effects include chloasma, melasma, rashes and urticaria. Erythema multiforme and erythema nodosum occur. Hypertension and thromboembolic disease are reported. Acute poisoning: Ingestion: Acute poisoning effects are mild and self limiting. Nausea, vomiting and break through vaginal bleeding have been reported following oral contraceptive overdose. Nasal congestion, visual disturbances, headache and hypertension have also been reported in association with estrogen overdose. ANIMAL/PLANT STUDIES: Relevant animal data: A correlation between the prolonged use of oral contraceptives and the development of liver cancer was demonstrated in rats. Induced DNA breaks in hamster kidneys, but not in livers, following 2 weeks of treatment with a single estradiol implant. Tumors of kidney, bone, testis, uterus and breast, were induced in animals exposed to estrogens. Mutagenicity: Estradiol induced DNA breaks in hamster renal cells, but not in hepatocytes.
Estrogens diffuse into their target cells and interact with a protein receptor. Target cells include the female reproductive tract, the mammary gland, the hypothalamus, and the pituitary. Estrogens increase the hepatic synthesis of sex hormone binding globulin (SHBG), thyroid-binding globulin (TBG), and other serum proteins and suppress follicle-stimulating hormone (FSH) from the anterior pituitary. This cascade is initiated by initially binding to the estrogen receptors. The combination of an estrogen with a progestin suppresses the hypothalamic-pituitary system, decreasing the secretion of gonadotropin-releasing hormone (GnRH).
Evaluation: There is sufficient evidence in humans for the carcinogenicity of post-menopausal estrogen therapy. There is sufficient evidence in experimental animals for the carcinogenicity of estradiol and estrone. There is limited evidence in experimental animals for the carcinogenicity of conjugated equine estrogens, equilin and estriol. There is inadequate evidence in experimental animals for the carcinogenicity of d-equilenin. Overall evaluation: Post-menopausal estrogen therapy is carcinogenic to humans (Group 1). /Post-menopausal estrogen therapy/
来源:Hazardous Substances Data Bank (HSDB)
毒理性
致癌物分类
对人类不具有致癌性(未被国际癌症研究机构IARC列名)。
No indication of carcinogenicity to humans (not listed by IARC).
◉ Summary of Use during Lactation:This record contains information specific to ethinyl estradiol used alone. Users with an interest in an oral contraceptive should consult the record entitled, "Contraceptives, Oral, Combined."
There is little information available on the use of ethinyl estradiol alone during breastfeeding. Levels in milk appear to be low. Based on studies on oral contraceptives that contain ethinyl estradiol, immediate side effects such as breast enlargement appear to occur rarely. It seems likely that doses of 30 mcg daily or greater can suppress lactation. The magnitude of the effect on lactation likely depends on the dose and the time of introduction postpartum, but data are not adequate to accurately define these doses and times.
◉ Effects in Breastfed Infants:Published information was not found as of the revision date on the effects of ethinyl estradiol alone on breastfed infants. However, case reports exist of breast enlargement in the infants of mothers taking combination oral contraceptives that contained ethinyl estradiol or its prodrug, mestranol.
◉ Effects on Lactation and Breastmilk:Published information was not found as of the revision date on the effects of ethinyl estradiol on milk production. However, numerous studies on combination contraceptives containing ethinyl estradiol or its prodrug mestranol indicate that doses of 30 mcg daily or greater might interfere with lactation. One study that used a contraceptive containing 10 mcg of ethinyl estradiol found no effect on lactation.
A retrospective cohort study compared 371 women who received high-dose estrogen (either 3 mg of diethylstilbestrol or 150 mcg of ethinyl estradiol daily)during adolescence for adult height reduction to 409 women who did not receive estrogen. No difference in breastfeeding duration was found between the two groups, indicating that high-dose estrogen during adolescence has no effect on later breastfeeding.
A 30µg oral dose of ethinylestradiol reaches a Cmax of 74.1±35.6pg/mL, with a Tmax of 1.5±0.5h, and an AUC of 487.4±166.6pg\*h/mL. A 1.2mg dose delivered via a patch reaches a Cmax of 28.8±10.3pg/mL, with a Tmax of 86±31h, and an AUC of3895±1423pg\*h/mL.
Ethinylestradiol is 59.2% eliminated in the urine and bile, while 2-3% is eliminated in the feces. Over 90% of ethinylestradiol is eliminated as the unchanged parent drug.
A 30µg oral dose has an apparent volume of distribution of 625.3±228.7L and a 1.2mg topical dose has an apparent volume of distribution of 11745.3±15934.8L.
Ethinylestradiol has an intravenous clearance of 16.47L/h, and an estimated renal clearance of approximately 2.1L/h. A 30µg oral dose has a clearance of 58.0±19.8L/h and a 1.2mg topical dose has a clearance of 303.5±100.5L/h.
Ethinyl estradiol is rapidly and almost completely absorbed. When the lowest and highest tablet strengths, 0.100 mg desogestrel/0.025 mg ethinyl estradiol and 0.150 mg desogestrel/0.025 mg ethinyl estradiol, were compared to solution, the relative bioavailability of ethinyl estradiol was 92% and 98%, respectively.
Synthesis and inhibition properties of a series of pyranose derivatives towards a Zn-metalloproteinase from Saccharomonospora canescens
摘要:
The Zn-proteinase, isolated from Saccharomonospora canescens (NPS), shares many common features with thermolysin, but considerable differences are also evident, as far as the substrate recognition site is concerned. In substrates of general structure AcylAlaAlaPhe 4NA, this neutral proteinase cleaves only the arylamide bond (non-typical activity of Zn-proteinases), while thermolysin attacks the peptide bond Ala-Phe. Phosphoramidon is a powerful tight binding inhibitor for thermolysin and significantly less specific towards NPS. The K-i-values (65 mu M for NPS vs 0.034 mu M for thermolysin) differ nearly 2000-folds. This implies significant differences in the specificity of the corresponding subsites. The carbohydrate moiety is supposed to accommodate in the S-1-subsite and the series of arabinopyranosides and glucopyranosides (12 compounds), which are assayed as inhibitors in a model system: NPS with SucAlaAlaPhe4NA as a substrate could be considered as mapping the S-1-subsite of NPS. Members of the series with an additional ring (3,4-epithio, 3,4-anhydro-derivatives) turned out to be reasonably good competitive inhibitors (K-i approximate to 0.1-0.2 mM are of the same order as the value for phosphoramidon). The structure of these compounds (8, 9, 11 and 12) seems to fit the size of the S-1-subsite and due to an appropriately oriented OH-group in addition, to protect the active site Zn2+. (C) 2010 Published by Elsevier Ltd.
Heterocyclic derivatives for the treatment of cancer and other proliferative diseases
申请人:——
公开号:US20020143182A1
公开(公告)日:2002-10-03
The invention relates to certain heterocyclic compounds useful for the treatment of cancer and other diseases, having the Formula (I):
1
wherein:
(a) m is an integer 0 or 1;
(b) R
12
is an alkyl, a substituted alkyl, a cycloalkyl, a substituted cycloalkyl, a heterocyclic, a substituted heterocyclic, a heteroaryl, a substituted heteroaryl, an aryl or a substituted aryl residue;
(c) Ar
3
is an aryl, a substituted aryl, a heteroaryl or a substituted heteroaryl residue;
(d) Ar
4
is an aryl, a substituted aryl, a heteroaryl or a substituted heteroaryl residue;
(e) R
5
is hydrogen, hydroxy, alkyl or substituted alkyl;
(f) - - - - - represents a bond present or absent; and
(g) W, X, Y and Z are independently or together C(O)—, C(S), S, O, or NH; or a pharmaceutically acceptable salt thereof.
The present invention provides a cobalamin-drug conjugate suitable for the treatment of tumor related diseases. Cobalamin is indirectly covalently bound to an anti-tumor drug via a cleavable linker and one or more optional spacers. Cobalamin is covalently bound to a first spacer or the cleavable linker via the 5′-OH of the cobalamin ribose ring. The drug is bound to a second spacer of the cleavable linker via an existing or added functional group on the drug. After administration, the conjugate forms a complex with transcobalamin (any of its isoforms). The complex then binds to a receptor on a cell membrane and is taken up into the cell. Once in the cell, an intracellular enzyme cleaves the conjugate thereby releasing the drug. Depending upon the structure of the conjugate, a particular class or type of intracellular enzyme affects the cleavage. Due to the high demand for cobalamin in growing cells, tumor cells typically take up a higher percentage of the conjugate than do normal non-growing cells. The conjugate of the invention advantageously provides a reduced systemic toxicity and enhanced efficacy as compared to a corresponding free drug.
[EN] INHIBITORS OF BRUTON'S TYROSINE KINASE<br/>[FR] INHIBITEURS DE TYROSINE KINASE DE BRUTON
申请人:BIOCAD JOINT STOCK CO
公开号:WO2018092047A1
公开(公告)日:2018-05-24
The present invention relates to a new compound of formula I: or pharmaceutically acceptable salt, solvate or stereoisomer thereof, wherein: V1 is C or N, V2 is C(R2) or N, whereby if V1 is C then V2 is N, if V1 is C then V2 is C(R2), or if V1 is N then V2 is C(R2); each n, k is independently 0, 1; each R2, R11 is independently H, D, Hal, CN, NR'R", C(O)NR'R", C1-C6 alkoxy; R3 is H, D, hydroxy, C(O)C1-C6 alkyl, C(O)C2-C6 alkenyl, C(O)C2-C6 alkynyl, C1-C6 alkyl; R4 is H, Hal, CN, CONR'R", hydroxy, C1-C6 alkyl, C1-C6 alkoxy; L is CH2, NH, O or chemical bond; R1 is selected from the group of the fragments, comprising: Fragment 1, Fragment 2, Fragment 3 each A1, A2, A3, A4 is independently CH, N, CHal; each A5, A6, A7, A8, A9 is independently C, CH or N; R5 is H, CN, Hal, CONR'R", C1-C6 alkyl, non-substituted or substituted by one or more halogens; each R' and R" is independently selected from the group, comprising H, C1-C6 alkyl, C1-C6 cycloalkyl, aryl; R6 is selected from the group: [formula II] each R7, R8, R9, R10 is independently vinyl, methylacetylenyl; Hal is CI, Br, I, F, which have properties of inhibitor of Bruton's tyrosine kinase (Btk), to pharmaceutical compositions containing such compounds, and their use as pharmaceuticals for treatment of diseases and disorder.
[EN] BRUTON'S TYROSINE KINASE INHIBITORS<br/>[FR] INHIBITEURS DE LA TYROSINE KINASE DE BRUTON
申请人:PFIZER
公开号:WO2014068527A1
公开(公告)日:2014-05-08
Disclosed herein are compounds that form covalent bonds with Bruton's tyrosine kinase (BTK). Methods for the preparation of the compounds are disclosed. Also disclosed are pharmaceutical compositions that include the compounds. Methods of using the BTK inhibitors are disclosed, alone or in combination with other therapeutic agents, for the treatment of autoimmune diseases or conditions, heteroimmune diseases or conditions, cancer, including lymphoma, and inflammatory diseases or conditions. (Formula I)
[EN] PROCESSES FOR MAKING TRIAZOLO[4,5D] PYRAMIDINE DERIVATIVES AND INTERMEDIATES THEREOF<br/>[FR] PROCÉDÉS DE PREPARATION DE DÉRIVÉS DE TRIAZOLO [4,5 D] PYRIMIDINE ET INTERMÉDIAIRES DE CEUX-CI
申请人:CORVUS PHARMACEUTICALS INC
公开号:WO2018183965A1
公开(公告)日:2018-10-04
Provided herein are, inter alia, methods for making triazolo[4,5]pyramidine derivatives and intermediates thereof that are useful for treating diseases.