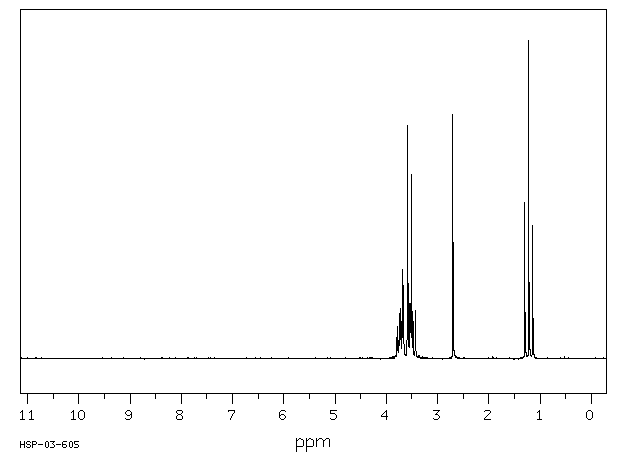
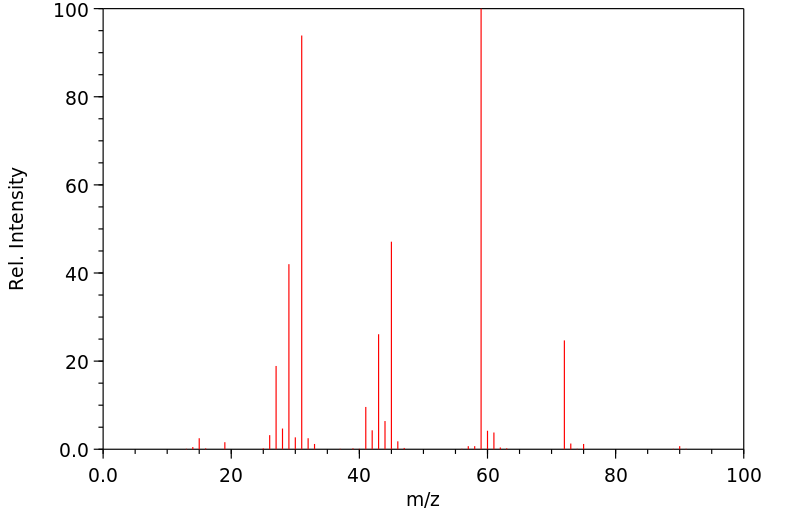
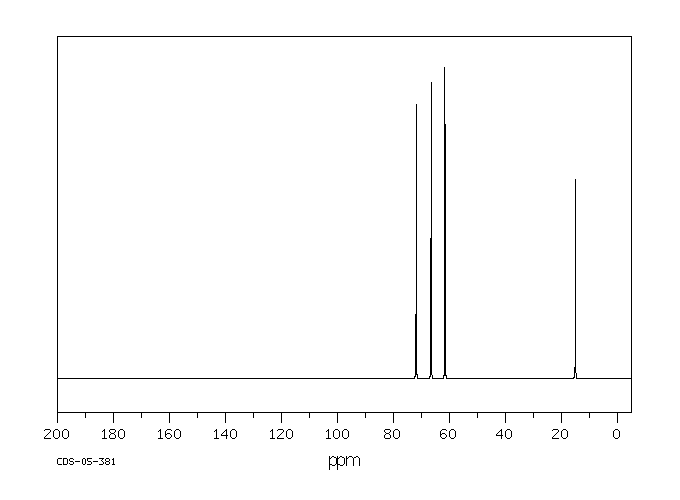
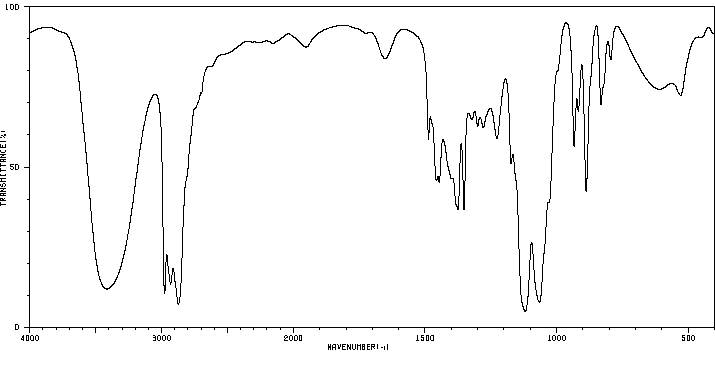
Male rats were given a single oral dose of ethylene glycol monoethyl ether, the dose ranging from plausible human exposures (0.5-1 mg/kg) to doses reported in the literature (100 mg/kg). Urinary excretion of ethoxyacetic acid and its glycine conjugate was followed up to 60 hr after dosing and compared to data of experimentally exposed human volunteers. In rats, the mean elimination half-life of free as well as conjugated ethoxyacetic acid was 7.2 hr for all doses. Ethoxyacetic acid was excreted partly as a glycine conjugate (on average 27%), the extent of conjugation being independent of the dose. The relative amount of ethylene glycol monoethyl ether recovered in urine as ethoxyacetic acid was only 13.4% for the lowest dose, but increased as the administered dose of ethylene-glycol-monoethyl-ether was higher, indicating that ethylene glycol monoethyl ether was metabolized at least in two parallel pathways of which one pathway becomes saturated at relatively low doses. In man, urinary excretion of ethoxyacetic acid for equivalent low doses of ethylene glycol monoethyl ether differed from that in the rat by a longer elimination half-life (mean 42 hr), by the absence of ethoxyacetic acid conjugates and by a higher recovery.
Ten healthy male subjects were exposed to ethylene glycol monoethyl ether and their urinary excretion of ethoxyacetic acid was followed up for 42 hours. Maximal excretion of ethoxyacetic acid was reached 3-4 hours after the end of the 4 hr exposure period. Afterwards, ethoxyacetic acid excretion declined slowly with a biological half-life of 21-24 hours. Ethoxyacetic acid excretion increased as the uptake of ethylene glycol monoethyl ether increased as a consequence of higher exposure concentration or pulmonary ventilation rate during physical exercise. On average, 23.1% of ethylene glycol monoethyl ether was recovered as ethoxyacetic acid within 42 hours. Quantitative relations between ethoxyacetic acid excretion and ethylene glycol monoethyl ether uptake were obtained.
/A group of/ 17 persons who were exposed to glycol ethers in a varnish production plant, were examined according to their external and internal solvent exposure. The workers in the production plant (n= 12) were exposed to average concentrations of ethoxyethanol, ethoxyethyl acetate, butoxyethanol, 1-methoxypropanol-2, 2-methoxypropyl-1-acetate and xylene of 2.8; 2.7; 1.1; 7.0; 2.8 and 1.7 ppm. Internal exposure was estimated by measuring butoxyethanol in blood as well as ethoxyacetic acid and butoxyacetic acid in urine samples. As expected, the highest values were found in the varnish production. The average post shift concentrations of butoxyethanol, ethoxyacetic acid and butoxyacetic acid were 121.3 ug/L; 167.8 and 10.5 mg/L. The relatively high concentrations of ethoxyacetic acid and butoxyacetic acid in pre-shift samples can be explained by the long half-lives of these metabolites. Most of the glycolethers were taken up through the skin. The authors think that a future tolerable limit value for the concentration of ethoxyacetic acid in urine should be in the order of 100 to 200 mg/L.
... Pathways and relative rates of metabolism of ethylene glycol monoethyl ether (EGEE) /were developed/ using rat and human hepatocytes. The concentrations of ethylene glycol monoethyl ether used were 0.02, 0.2, 2.0, and 10.0 mM. Metabolites were analyzed by HPLC. Ethylene glycol (EG) was the major metabolite of EGEE (30%). The percentage of EGEE converted to ethoxyacetic acid (EAA) in rat hepatocytes was similar at all concentrations ... The Vmax value for the conversion of EGEE to ethoxyacetic acid with rat hepatocytes was similar to those obtained for the conversion of ethylene glycol monomethyl ether (EGME) to /methoxyacetic acid / (MAA) and ethylene glycol monobutyl ether (EGBE) to butoxyacetic acid (BAA). In human hepatocytes, Vmax followed the order of EGBE>EGEE>EGME. Vmax was 15-20 fold higher in rat than in human hepatocytes.
IDENTIFICATION AND USE: Ethylene Glycol Monoethyl ether (EGEE) is a colorless liquid with mild, agreeable odor. It is used as a solvent for nitrocellulose, lacquers and dopes; in varnish removers, cleansing solutions, dye baths; finishing leather with water pigments and dye solutions; increasing stability of emulsions. HUMAN EXPOSURE AND TOXICITY: Short-term exposure: The substance is mildly irritating to the eyes and respiratory tract, may cause effects on the central nervous system, blood, bone marrow, kidneys and liver. Exposure at high levels could cause unconsciousness. Long-term or repeated exposure: The liquid defats the skin, may have effects on the blood and bone marrow. The major long-term effects of exposure for humans are developmental, testicular, and hematological toxicity. A cross-sectional evaluation of semen quality was carried out in workers exposed to EGEE. A control group of workers from the same plant was included. Workers exposed to EGEE had significantly lower average sperm counts than controls, although both groups had lower sperm counts than those found in other occupational groups. In other study, hemoglobin, hematocrit, red cell indices, total and differential white blood cell counts, and platelet count were measured in shipyard painters and control subjects as part of a cross-sectional, observational study of the effects of ethylene glycol ethers. A significant proportion of painters were anemic (10%) and granulocytopenic (5%); none of the controls were affected. ANIMAL TOXICITY STUDIES: EGEE administered sc for 4 weeks at doses of up to 0.38 g/kg/day caused no deaths in rats. Dose levels of 0.185 and 0.38 g/kg caused dyspnea, somnolence, mild ataxia, some growth depression in females, and reduction of hemoglobin levels and hematocrit values. At the 0.38 g/kg level interstitial testicular edema, dissociation of liver parenchyma and tubular lesions of the kidney were observed. The effects of EGEE on testicular cell populations in pubertal and adult male rats were investigated. In adult rats, the treatment of EGEE at the dose of 400 mg/kg bw decreased significantly the populations of haploid cells, while it increased those of diploid and tetraploid cells. In pubertal rats, the treatment of EGEE at the dose of 400 mg/kg bw caused only minimal changes in the relative percent of testicular cell types. Thus the effects of EGEE on testicular function in pubertal rats appear to be less pronounced than in adult rats. Developmental studies utilizing inhalation exposure of rats and rabbits during gestation were conducted with EGEE. It was strongly embryotoxic at the higher exposure levels employed and was teratogenic at the lower concentration. In other study, mice were intubated during gestation and were evaluated for signs of EGEE toxicity. EGEE produced embryo lethality and malformations, and decreased fetal weight at a dose level which was not maternally toxic in the teratology probe. In the postnatal study, EGEE decreased litter size and neonatal body weight, while litter size continued to decrease beyond neonatal period. Body weights of surviving pups were not significantly different from control. Pups exposed prenatally to EGEE developed kinked tail which was not apparent in fetuses or neonates. Histologic studies on the effects of EGEE identified dividing spermatocyte as a primary target cell type in the testis. Further studies were undertaken to assess possible effects of EGEE on late-stage and epididymal spermatids, and spermatogonia. Adult male rats were dosed orally with EGEE up to 200 mg /kg/day for 5 days. Each male was then mated with two females weekly for 8 weeks. The fertility of males treated with 200 mg EGEE/kg declined at week 4, and remained low. There was a decrease in the number of litters sired at week 5 after dosing in the 100 mg EGEE/kg group. There were time- and dose-related decreases in sperm concentration and motility, primarily in the 100 and 200 mg/kg groups, as well as concurrent elevations in the number of abnormal sperm forms in the epididymis. Behavioral and neurochemical alterations were seen in the offspring of rats exposed to EGEE. EGEE was positive in cultured Chinese hamster ovary cells for the induction of chromosomal aberrations and sister-chromatid exchanges. EGEE was negative in the Salmonella and mouse lymphoma assays and in the Drosophila test for sex linked recessive lethal mutations. ECOTOXICITY STUDIES: The acute toxicity of EGEE is very low for arthropods (LC50 > 4 g/L) and for freshwater fish (LC50 > 10 g/L).
来源:Hazardous Substances Data Bank (HSDB)
毒理性
致癌物分类
对人类不具有致癌性(未被国际癌症研究机构IARC列名)。
No indication of carcinogenicity to humans (not listed by IARC).
来源:Toxin and Toxin Target Database (T3DB)
毒理性
暴露途径
这种物质可以通过吸入、皮肤接触和摄入被身体吸收。
The substance can be absorbed into the body by inhalation, through the skin and by ingestion.
来源:ILO-WHO International Chemical Safety Cards (ICSCs)
The urinary excretion of ethoxyacetic acid (EAA) was studied in a group of five women daily exposed to the ethyl ether of ethylene glycol (EGEE) and the ethyl ether of ethylene glycol acetate (EGEE-Ac) during 5 day of normal production and 7 day after a 12 day production stop. The mean combined exposure concentration of EGEE and EGEE-Ac (expressed in equivalent weight of EGEE) was 14.0 mg/cu m with occasional slight excursions above the current Belgian occupational exposure limit. The daily combined exposure profiles for EGEE and EGEE-Ac were rather constant during the first observation period, but they tended to decrease during the last week. The urinary EAA excretion clearly increased during the work week. Over the weekends the elimination was far from complete, and even after a prolonged nonexposure period of 12 day traces of the metabolite were still detectable. Based on the observations from the first period, a good linear correlation (r = 0.92) was found between the average exposure over 5 day (14.4 mg/cu m) and the EAA excretion at the end of the week (105.7 mg/g creatinine). An EAA estimate of 150 +/- 35 mg/g was found to correspond with repeated 5 day full-shift exposures to the respective occupational exposure limit of EGEE (19 mg/cu m) or EGEE-Ac (27 mg/cu m).
Percutaneous absorption and cutaneous metabolism of 2-ethoxyethanol were assessed in vivo and with an in vitro flow-through diffusion system. Topical application of undiluted (14)C-ethoxyethanol to occluded rat skin in vivo resulted in 25% of the dose being absorbed after 24 hr. The major routes of excretion included the urine (15%), expiration as carbon dioxide (6%), and feces (1.2%), while little of the dose remained in the carcass (1.3%). Free ethoxyethanol, ethoxyacetic acid, and glycine conjugate were detected in urine. Permeation rates of ethoxyethanol through unoccluded rat split skin (20%) were greater than rat whole skin (11%), while absorption through human split skin (8%) was lower than the rat. Absorption of undiluted ethoxyethanol through occluded rat split skin in vitro (22%) most accurately predicted absorption through rat skin in vivo. However, ethoxyethanol absorption (29%) was enhanced by application in methanol. First pass metabolism of ethoxyethanol was not detected during percutaneous penetration through viable human or rat skin in vitro or rat skin in vivo. However, rat skin cytosol had the potential to metabolize ethoxyethanol, suggesting that the rapid penetration through skin in vivo prevented metabolism and that systemic exposure after skin contact with 2-ethoxyethanol is likely to be to the parent compound. In conclusion, the in vitro system provided a reasonable estimate of dermal absorption for the rat in vivo and comparison of human and rat skin in vitro indicated 2-ethoxyethanol absorption in humans is about one-third of that in the rat.
Percutaneous absorption of /2-ethoxyethanol/ EE, /2-butoxyethanol/ BE and /1-methoxy-2-propanol/ M2P, in aqueous solution (3 mg/mL, 200 microL) or undiluted (10.5 microL), though full thickness or dermatomed human breast skin (0.64 sq cm exposed area) was measured for 24 hr using flow-through diffusion cells. Tissue culture medium was used as receptor fluid, with 2% (w/v) bovine serum albumin (BSA) or 2%-6% (w/v) polyethylene glycol 20 oleyl ether (PEG 20) added for some studies. Volatilised test compounds were trapped on charcoal filters placed above cells. RESULTS: In aqueous solution, steady-state flux of BE (544+/-64 nmol/sq cm/hr) exceeded that of EE (143+/-19 nmol/sq cm/hr) and M2P (48+/-6 nmol/sq cm/hr). Reducing the dose volume to 100 microL decreased the steady-state flux of BE by about 55%, though the flux of EE was approximately doubled. Doubling the dose concentration of EE increased the flux by about eight-fold. Using full thickness skin increased tau of both EE and BE and reduced the steady-state flux of BE. Absorption rates of undiluted solvents in finite doses exceeded those measured with aqueous solutions, though the apparent permeability coefficient was higher with aqueous doses. Addition of BSA or PEG 20 to receptor fluid markedly increased absorption in both aqueous and undiluted doses. The dermal absorption potential of M2P from a liquid application was markedly lower than from EE or BE in all but infinite undiluted doses. The influence of receptor fluid on dermal absorption of glycol ethers could be relevant to prediction of absorption in vivo.
Studies to evaluate the rate of its elimination were conducted in rats using a priming dose of 140 mg/kg iv followed by the infusion initially of 8, then of 16 mg/kg/min. Under these conditions it was found that this product was excreted via the lung at a rate of slightly more than 8 mg/kg/min unchanged.
[EN] COMPOSITIONS, SYNTHESIS, AND METHODS OF USING PHENYLCYCLOALKYLMETHYLAMINE DERIVATIVES [FR] COMPOSITIONS, SYNTHÈSES ET PROCÉDÉS D'UTILISATION DE DÉRIVÉS DE PHÉNYLCYCLOALKYLMÉTHYLAMINE
The present invention relates compounds of formula (I)
wherein A and R
1
are as defined in the specification, pharmaceutical compositions comprising such compounds, and methods of treating conditions and disorders using such compounds and pharmaceutical compositions.
Disclosed herein are compounds of formula (I)
wherein Ring A and R
1
are as defined in the specification. Pharmaceutical compositions comprising such compounds, and methods for treating conditions and disorders using such compounds and pharmaceutical compositions are also disclosed.
Synthesis, antiproliferative activity and molecular docking of thiocolchicine urethanes
作者:Urszula Majcher、Alicja Urbaniak、Ewa Maj、Mahshad Moshari、Magdalena Delgado、Joanna Wietrzyk、Franz Bartl、Timothy C. Chambers、Jack A. Tuszynski、Adam Huczyński
DOI:10.1016/j.bioorg.2018.09.004
日期:2018.12
colchicine exert their therapeutic effect by changing the dynamics of tubulin and its polymer form, microtubules. The identification of tubulin as a potentialtarget for anticancer drugs has led to extensive research followed by clinical development of numerous compounds from several families. In this paper we report on the design, synthesis and in vitro evaluation of a group of thiocolchicine derivatives
The cobalt-catalyzed regioselective C-H alkoxylation of 1-naphthylamide with diols through a bidentate-chelation assistance has been developed. In this transformation, not only linear diols, but also branched diols and oligoethylene glycols were tolerated under current reaction conditions, affording the corresponding hydroxyalkyl aryl ethers. In addition, control experiments suggested that picolinoyl
The present invention relates to compounds or pharmaceutically-acceptable salts thereof, processes for preparing them, pharmaceutical compositions containing them and their use in therapy. The invention particularly relates to compounds that are polo-like kinase (PLKs) inhibitors useful for the treatment of disease states mediated by PLK, especially PLK4, in particular such compounds that are useful in the treatment of pathological processes which involve an aberrant cellular proliferation, such as tumour growth, rheumatoid arthritis, restenosis and atherosclerosis.