
烟酰甘氨酸 | 583-08-4
物质功能分类
中文名称
烟酰甘氨酸
中文别名
烟尿酸;氮-烟酰甘胺酸;2-[(3-吡啶羰基)氨基]乙酸
英文名称
nicotinoylglycine
英文别名
Nicotinuric acid;N-nicotinoyl glycine;Nicotinursaeure;2-(pyridine-3-carbonylamino)acetic acid

CAS
583-08-4
化学式
C8H8N2O3
mdl
MFCD00023578
分子量
180.163
InChiKey
ZBSGKPYXQINNGF-UHFFFAOYSA-N
BEILSTEIN
——
EINECS
——
-
物化性质
-
计算性质
-
ADMET
-
安全信息
-
SDS
-
制备方法与用途
-
上下游信息
-
文献信息
-
表征谱图
-
同类化合物
-
相关功能分类
-
相关结构分类
物化性质
-
熔点:256-2580C (dec.)
-
沸点:85-90 °C(Press: 12-20 Torr)
-
密度:1.49 g/cm3
-
溶解度:碱性溶液(微溶)、DMSO(微溶)、甲醇(微溶、加热、超声处理)
-
物理描述:Solid
-
碰撞截面:143.6 Ų [M+H]+ [CCS Type: DT, Method: single field calibrated with Agilent tune mix (Agilent)]
计算性质
-
辛醇/水分配系数(LogP):-0.4
-
重原子数:13
-
可旋转键数:3
-
环数:1.0
-
sp3杂化的碳原子比例:0.125
-
拓扑面积:79.3
-
氢给体数:2
-
氢受体数:4
安全信息
-
危险等级:IRRITANT
-
危险品标志:Xi
-
WGK Germany:3
-
海关编码:2933399090
-
危险性防范说明:P261,P305+P351+P338
-
危险性描述:H302,H315,H319,H335
-
储存条件:存储于0-5°C环境中
SDS
模块 1. 化学品
1.1 产品标识符
: Nicotinuric acid
产品名称
1.2 鉴别的其他方法
Nicotinylglycine
1.3 有关的确定了的物质或混合物的用途和建议不适合的用途
仅供科研用途,不作为药物、家庭备用药或其它用途。
模块 2. 危险性概述
2.1 GHS分类
根据化学品全球统一分类与标签制度(GHS)的规定,不是危险物质或混合物。
2.3 其它危害物 - 无
模块 3. 成分/组成信息
3.1 物 质
: Nicotinylglycine
别名
: C8H8N2O3
分子式
: 180.16 g/mol
分子量
无
模块 4. 急救措施
4.1 必要的急救措施描述
吸入
如果吸入,请将患者移到新鲜空气处。 如果停止了呼吸,给于人工呼吸。
皮肤接触
用肥皂和大量的水冲洗。
眼睛接触
用水冲洗眼睛作为预防措施。
食入
切勿给失去知觉者从嘴里喂食任何东西。 用水漱口。
4.2 主要症状和影响,急性和迟发效应
据我们所知,此化学,物理和毒性性质尚未经完整的研究。
4.3 及时的医疗处理和所需的特殊处理的说明和指示
无数据资料
模块 5. 消防措施
5.1 灭火介质
灭火方法及灭火剂
用水雾,耐醇泡沫,干粉或二氧化碳灭火。
5.2 源于此物质或混合物的特别的危害
碳氧化物, 氮氧化物
5.3 给消防员的建议
如必要的话,戴自给式呼吸器去救火。
5.4 进一步信息
无数据资料
模块 6. 泄露应急处理
6.1 人员的预防,防护设备和紧急处理程序
防止粉尘的生成。 防止吸入蒸汽、气雾或气体。
6.2 环境保护措施
不要让产物进入下水道。
6.3 抑制和清除溢出物的方法和材料
扫掉和铲掉。 存放进适当的闭口容器中待处理。
6.4 参考其他部分
丢弃处理请参阅第13节。
模块 7. 操作处置与储存
7.1 安全操作的注意事项
在有粉尘生成的地方,提供合适的排风设备。一般性的防火保护措施。
7.2 安全储存的条件,包括任何不兼容性
贮存在阴凉处。 容器保持紧闭,储存在干燥通风处。
7.3 特定用途
无数据资料
模块 8. 接触控制和个体防护
8.1 容许浓度
最高容许浓度
没有已知的国家规定的暴露极限。
8.2 暴露控制
适当的技术控制
常规的工业卫生操作。
个体防护设备
眼/面保护
请使用经官方标准如NIOSH (美国) 或 EN 166(欧盟) 检测与批准的设备防护眼部。
皮肤保护
戴手套取 手套在使用前必须受检查。
请使用合适的方法脱除手套(不要接触手套外部表面),避免任何皮肤部位接触此产品.
使用后请将被污染过的手套根据相关法律法规和有效的实验室规章程序谨慎处理. 请清洗并吹干双手
所选择的保护手套必须符合EU的89/686/EEC规定和从它衍生出来的EN 376标准。
身体保护
根据危险物质的类型,浓度和量,以及特定的工作场所来选择人体保护措施。,
防护设备的类型必须根据特定工作场所中的危险物的浓度和含量来选择。
呼吸系统防护
不需要保护呼吸。如需防护粉尘损害,请使用N95型(US)或P1型(EN 143)防尘面具。
呼吸器使用经过测试并通过政府标准如NIOSH(US)或CEN(EU)的呼吸器和零件。
模块 9. 理化特性
9.1 基本的理化特性的信息
a) 外观与性状
形状: 固体
b) 气味
无数据资料
c) 气味阈值
无数据资料
d) pH值
无数据资料
e) 熔点/凝固点
无数据资料
f) 起始沸点和沸程
无数据资料
g) 闪点
无数据资料
h) 蒸发速率
无数据资料
i) 易燃性(固体,气体)
无数据资料
j) 高的/低的燃烧性或爆炸性限度 无数据资料
k) 蒸汽压
无数据资料
l) 蒸汽密度
无数据资料
m) 相对密度
无数据资料
n) 水溶性
无数据资料
o) n-辛醇/水分配系数
无数据资料
p) 自燃温度
无数据资料
q) 分解温度
无数据资料
r) 粘度
无数据资料
模块 10. 稳定性和反应活性
10.1 反应性
无数据资料
10.2 稳定性
无数据资料
10.3 危险反应的可能性
无数据资料
10.4 应避免的条件
无数据资料
10.5 不兼容的材料
无数据资料
10.6 危险的分解产物
其它分解产物 - 无数据资料
模块 11. 毒理学资料
11.1 毒理学影响的信息
急性毒性
无数据资料
皮肤刺激或腐蚀
无数据资料
眼睛刺激或腐蚀
无数据资料
呼吸道或皮肤过敏
无数据资料
生殖细胞突变性
无数据资料
致癌性
IARC:
此产品中没有大于或等于 0。1%含量的组分被 IARC鉴别为可能的或肯定的人类致癌物。
生殖毒性
无数据资料
特异性靶器官系统毒性(一次接触)
无数据资料
特异性靶器官系统毒性(反复接触)
无数据资料
吸入危险
无数据资料
潜在的健康影响
吸入 吸入可能有害。 可能引起呼吸道刺激。
摄入 如服入是有害的。
皮肤 如果通过皮肤吸收可能是有害的。 可能引起皮肤刺激。
眼睛 可能引起眼睛刺激。
接触后的征兆和症状
据我们所知,此化学,物理和毒性性质尚未经完整的研究。
附加说明
化学物质毒性作用登记: 无数据资料
模块 12. 生态学资料
12.1 生态毒性
无数据资料
12.2 持久存留性和降解性
无数据资料
12.3 潜在的生物蓄积性
无数据资料
12.4 土壤中的迁移性
无数据资料
12.5 PBT 和 vPvB的结果评价
无数据资料
12.6 其它不利的影响
无数据资料
模块 13. 废弃处置
13.1 废物处理方法
产品
将剩余的和未回收的溶液交给处理公司。
受污染的容器和包装
作为未用过的产品弃置。
模块 14. 运输信息
14.1 联合国危险货物编号
欧洲陆运危规: - 国际海运危规: - 国际空运危规: -
14.2 联合国(UN)规定的名称
欧洲陆运危规: 非危险货物
国际海运危规: 非危险货物
国际空运危规: 非危险货物
14.3 运输危险类别
欧洲陆运危规: - 国际海运危规: - 国际空运危规: -
14.4 包裹组
欧洲陆运危规: - 国际海运危规: - 国际空运危规: -
14.5 环境危险
欧洲陆运危规: 否 国际海运危规 海运污染物: 否 国际空运危规: 否
14.6 对使用者的特别提醒
无数据资料
模块 15 - 法规信息
N/A
模块16 - 其他信息
N/A
制备方法与用途
上下游信息
-
上游原料
中文名称 英文名称 CAS号 化学式 分子量 (吡啶-3-羰基)-氨基乙酸乙酯 N3-((ethoxycarbonyl)-methyl)nicotinamide 54466-74-9 C10H12N2O3 208.217 —— nicotinyl-glycine-tert-butyl ester 1037820-84-0 C12H16N2O3 236.271 烟酰胺 nicotinamide 98-92-0 C6H6N2O 122.126 3-吡啶甲酰肼 Nicotinic acid hydrazide 553-53-7 C6H7N3O 137.141 -
下游产品
中文名称 英文名称 CAS号 化学式 分子量 烟酰胺 nicotinamide 98-92-0 C6H6N2O 122.126
反应信息
-
作为反应物:描述:参考文献:名称:Synthesis and Quantitative Structure-activity Relationships Study for Arylpropenamide Derivatives as Inhibitors of Hepatitis B Virus Replication摘要:A series of new arylpropenamide derivatives containing different aryl groups were synthesized, characterized, and evaluated for their anti‐hepatitis B virus (DOI:10.1111/cbdd.12774
-
作为产物:参考文献:名称:新型硫化氢释放烟酸衍生物的合成及生物评价摘要:合成了十二种新的缓释硫化氢供体ADT-OH与烟酸结合的杂种。所有结构均通过1 H NMR确证,131 H NMR和MS光谱。用MTT分析评估目标化合物对谷氨酸诱导的损伤对海马神经元HT22细胞的神经保护作用,并在100μM浓度对未经谷氨酰胺处理的HT22细胞的毒性。在永久性大脑中动脉闭塞(pMCAO)小鼠模型中,在缺血后3小时通过腹膜内注射进一步研究了该活性化合物对缺血性梗死体积的影响。结果表明,所有化合物在大多数实验浓度下均能显着保护HT22细胞免受谷氨酸诱导的损伤,并且在高浓度下对正常HT22细胞没有或只有很少的神经毒性。更重要的是,化合物A6在pMCAO模型中显着减少了梗塞体积。这些结果表明,化合物A6可能有望用于进一步评估对脑缺血损伤的干预。DOI:10.1016/j.bmc.2016.08.060
文献信息
-
Methods and compounds for inhibiting mrp1申请人:——公开号:US20030100576A1公开(公告)日:2003-05-29The present invention further relates to a method of inhibiting MRP1 in a mammal which comprises administering to a mammal in need thereof an effective amount of a compound of formula (I).本发明还涉及一种抑制哺乳动物中MRP1的方法,包括向需要的哺乳动物施用化合物(I)的有效量。
-
Novel cinnamaldehyde compound having an azido group申请人:——公开号:US20030191329A1公开(公告)日:2003-10-09The invention provides a novel azidocinnamaldehyde compound which, when used as an intermediate for providing a photosensitive moiety of a photoresist, introduces a photosensitive moiety having high sensitivity and attaining high contrast between the exposed portion and the unexposed portion, and which per se can serve as a photosensitive moiety. The azidocinnamaldehyde compound is represented by formula(I): 1 wherein R represents a lower alkyl group and Y represents hydrogen or a sulfonate salt group.
-
Hepatitis C virus polymerase inhibitors申请人:Blaney Jeffrey Mark公开号:US20080200512A1公开(公告)日:2008-08-21The invention provides a compound of formula: and methods for making and using the same, where R 1 and R 2 are independently hydrogen, C 1-6 alkyl or C 1-6 haloalkyl; R 3 is hydrogen, halogen, C 1-6 alkyl or C 1-6 alkoxy; R 4 is hydrogen or halogen; and R 5 is a mono- or di-amino acid moiety, a succinic acid moiety, a urea moiety, or a derivative thereof. The invention also provides pharmaceutical compositions comprising a compound of formula I, and methods for making and using the same.
-
Discovery of liver-targeted inhibitors of stearoyl-CoA desaturase (SCD1)作者:Yongqi Deng、Zhiwei Yang、Gerald W. Shipps、Sie-Mun Lo、Robert West、Joyce Hwa、Shuqin Zheng、Constance Farley、Jean Lachowicz、Margaret van Heek、Alan S. Bass、Dinesh P. Sinha、Craig R. Mahon、Mark E. CartwrightDOI:10.1016/j.bmcl.2012.11.075日期:2013.2Inhibitors based on a benzo-fused spirocyclic oxazepine scaffold were discovered for stearoyl-coenzyme A (CoA) desaturase 1 (SCD1) and subsequently optimized to potent compounds with favorable pharmacokinetic profiles and in vivo efficacy in reducing the desaturation index in a mouse model. Initial optimization revealed potency preferences for the oxazepine core and benzylic positions, while substituents发现基于苯并稠合的螺环型奥氮平骨架的抑制剂的硬脂酰辅酶A(CoA)去饱和酶1(SCD1),随后被优化为具有良好药代动力学特征的有效化合物,并具有降低小鼠模型中去饱和指数的体内功效。最初的优化显示了对奥氮平核心和苄基位置的效用偏好,而哌啶部分上的取代基则更具耐受性,并可以调节效价和PK特性。在制备和测试哌啶氮上的一系列官能团后,鉴定出三类具有一位数纳摩尔效能的类似物:甘氨酸酰胺,杂环连接的酰胺和噻唑。在回应有关目标定位和潜在的基于机制的副作用的担忧时,在可利用的大鼠PK模型中,还做出了初步努力来改善肝脏浓度。先进的化合物开发了17m分子,该分子具有5-羧基-2-噻唑亚结构,并附加到螺环哌啶骨架上,可以满足更详细研究的体外和体内要求。
-
[EN] PYRONE COMPOUND AND ITS USE FOR PEST CONTROL<br/>[FR] COMPOSÉ DE PYRONE ET SON UTILISATION POUR LUTTER CONTRE LES ORGANISMES NUISIBLES申请人:SUMITOMO CHEMICAL CO公开号:WO2011049150A1公开(公告)日:2011-04-28A pyrone compound represented by formula (1) has an excellent controlling effect on pests. Since the compound of formula (1) has a controlling activity on pests, the compound is useful as an active ingredient of a pest control agent.
表征谱图
-
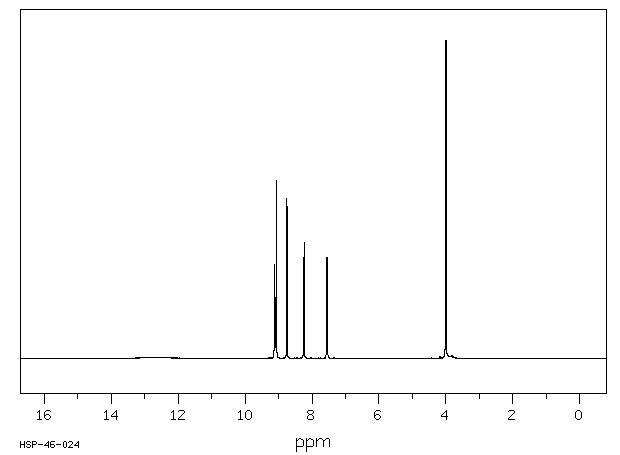
氢谱1HNMR
-
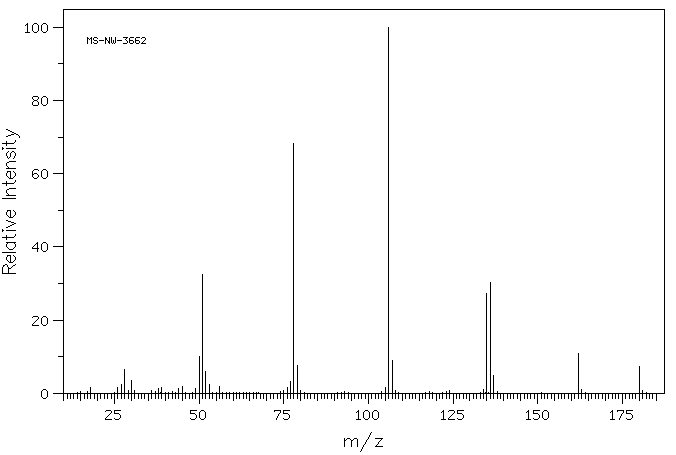
质谱MS
-
碳谱13CNMR
-
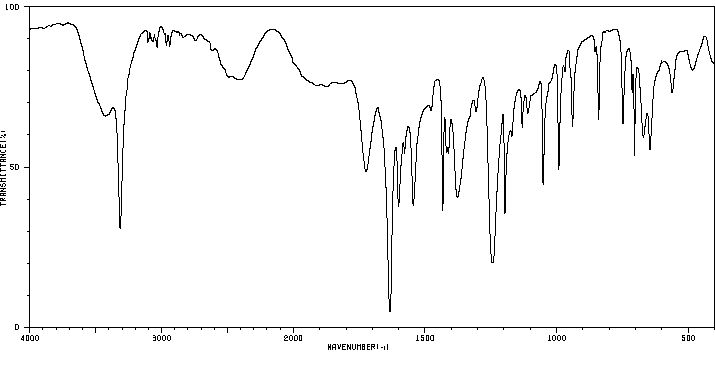
红外IR
-
拉曼Raman
-
峰位数据
-
峰位匹配
-
表征信息
同类化合物
(甲基3-(二甲基氨基)-2-苯基-2H-azirene-2-羧酸乙酯)
(±)-盐酸氯吡格雷
(±)-丙酰肉碱氯化物
(d(CH2)51,Tyr(Me)2,Arg8)-血管加压素
(S)-(+)-α-氨基-4-羧基-2-甲基苯乙酸
(S)-阿拉考特盐酸盐
(S)-赖诺普利-d5钠
(S)-2-氨基-5-氧代己酸,氢溴酸盐
(S)-2-[[[(1R,2R)-2-[[[3,5-双(叔丁基)-2-羟基苯基]亚甲基]氨基]环己基]硫脲基]-N-苄基-N,3,3-三甲基丁酰胺
(S)-2-[3-[(1R,2R)-2-(二丙基氨基)环己基]硫脲基]-N-异丙基-3,3-二甲基丁酰胺
(S)-1-(4-氨基氧基乙酰胺基苄基)乙二胺四乙酸
(S)-1-[N-[3-苯基-1-[(苯基甲氧基)羰基]丙基]-L-丙氨酰基]-L-脯氨酸
(R)-乙基N-甲酰基-N-(1-苯乙基)甘氨酸
(R)-丙酰肉碱-d3氯化物
(R)-4-N-Cbz-哌嗪-2-甲酸甲酯
(R)-3-氨基-2-苄基丙酸盐酸盐
(R)-1-(3-溴-2-甲基-1-氧丙基)-L-脯氨酸
(N-[(苄氧基)羰基]丙氨酰-N〜5〜-(diaminomethylidene)鸟氨酸)
(6-氯-2-吲哚基甲基)乙酰氨基丙二酸二乙酯
(4R)-N-亚硝基噻唑烷-4-羧酸
(3R)-1-噻-4-氮杂螺[4.4]壬烷-3-羧酸
(3-硝基-1H-1,2,4-三唑-1-基)乙酸乙酯
(2S,4R)-Boc-4-环己基-吡咯烷-2-羧酸
(2S,3S,5S)-2-氨基-3-羟基-1,6-二苯己烷-5-N-氨基甲酰基-L-缬氨酸
(2S,3S)-3-((S)-1-((1-(4-氟苯基)-1H-1,2,3-三唑-4-基)-甲基氨基)-1-氧-3-(噻唑-4-基)丙-2-基氨基甲酰基)-环氧乙烷-2-羧酸
(2S)-2,6-二氨基-N-[4-(5-氟-1,3-苯并噻唑-2-基)-2-甲基苯基]己酰胺二盐酸盐
(2S)-2-氨基-N,3,3-三甲基-N-(苯甲基)丁酰胺
(2S)-2-氨基-3-甲基-N-2-吡啶基丁酰胺
(2S)-2-氨基-3,3-二甲基-N-(苯基甲基)丁酰胺,
(2S)-2-氨基-3,3-二甲基-N-2-吡啶基丁酰胺
(2S,4R)-1-((S)-2-氨基-3,3-二甲基丁酰基)-4-羟基-N-(4-(4-甲基噻唑-5-基)苄基)吡咯烷-2-甲酰胺盐酸盐
(2R,3'S)苯那普利叔丁基酯d5
(2R)-2-氨基-3,3-二甲基-N-(苯甲基)丁酰胺
(2-氯丙烯基)草酰氯
(1S,3S,5S)-2-Boc-2-氮杂双环[3.1.0]己烷-3-羧酸
(1R,5R,6R)-5-(1-乙基丙氧基)-7-氧杂双环[4.1.0]庚-3-烯-3-羧酸乙基酯
(1R,4R,5S,6R)-4-氨基-2-氧杂双环[3.1.0]己烷-4,6-二羧酸
齐特巴坦
齐德巴坦钠盐
齐墩果-12-烯-28-酸,2,3-二羟基-,苯基甲基酯,(2a,3a)-
齐墩果-12-烯-28-酸,2,3-二羟基-,羧基甲基酯,(2a,3b)-(9CI)
黄酮-8-乙酸二甲氨基乙基酯
黄荧菌素
黄体生成激素释放激素(1-6)
黄体生成激素释放激素 (1-5) 酰肼
黄体瑞林
麦醇溶蛋白
麦角硫因
麦芽聚糖六乙酸酯
麦根酸
相关功能分类

联系我们



关注我们

公众号

