
3,4-dibenzyloxy-2-hydroxyacetophenone | 2652-27-9
中文名称
——
中文别名
——
英文名称
3,4-dibenzyloxy-2-hydroxyacetophenone
英文别名
1-(3,4-bis(benzyloxy)-2-hydroxyphenyl)ethanone;Ethanone, 1-[2-hydroxy-3,4-bis(phenylmethoxy)phenyl]-;1-[2-hydroxy-3,4-bis(phenylmethoxy)phenyl]ethanone

CAS
2652-27-9
化学式
C22H20O4
mdl
——
分子量
348.398
InChiKey
ACRPAJBIWLUSEP-UHFFFAOYSA-N
BEILSTEIN
——
EINECS
——
-
物化性质
-
计算性质
-
ADMET
-
安全信息
-
SDS
-
制备方法与用途
-
上下游信息
-
文献信息
-
表征谱图
-
同类化合物
-
相关功能分类
-
相关结构分类
计算性质
-
辛醇/水分配系数(LogP):4.7
-
重原子数:26
-
可旋转键数:7
-
环数:3.0
-
sp3杂化的碳原子比例:0.14
-
拓扑面积:55.8
-
氢给体数:1
-
氢受体数:4
上下游信息
-
上游原料
中文名称 英文名称 CAS号 化学式 分子量 —— 2,3,4-tribenzyloxyacetophenone 50439-63-9 C29H26O4 438.523 2',3',4'-三羟基苯乙酮 2',3',4'-trihydroxyacetophenone 528-21-2 C8H8O4 168.149 -
下游产品
中文名称 英文名称 CAS号 化学式 分子量 —— 3,4-dibenzyloxy-2,5-dihydroxyacetophenone 158148-87-9 C22H20O5 364.398 —— 3,4-dibenzyloxy-2-hydroxy-5-methoxyacetophenone 158148-88-0 C23H22O5 378.425 —— 3',4'-dibenzyloxy-2'-methoxyacetophenone 27829-92-1 C23H22O4 362.425 —— 1-[2-hydroxy-3,4-bis(phenylmethoxy)phenyl]-3-phenylprop-2-en-1-one 93827-33-9 C29H24O4 436.507 3,4-二羟基-2-甲氧基苯乙酮 3',4'-dihydroxy-2'-methoxyacetophenone 27829-93-2 C9H10O4 182.176
反应信息
-
作为反应物:描述:3,4-dibenzyloxy-2-hydroxyacetophenone 在 palladium 10% on activated carbon 、 氢气 、 碘 、 二甲基亚砜 、 potassium hydroxide 作用下, 以 四氢呋喃 、 甲醇 、 乙醇 、 水 为溶剂, 反应 51.0h, 生成 7,8,4'-Trihydroxyflavone参考文献:名称:藤黄酸(IV)的研究:探索与IκB激酶-β(IKKβ)的构效关系摘要:以前,我们已经报道了一系列藤黄酸的类似物,并鉴定出了一种具有与藤黄酸相当的体外生长抑制作用的化合物。但是,尚未鉴定其靶蛋白以及靶上的关键药效基序。在本文中,我们报道藤黄酸及其类似物通过抑制TNFα/NF-κB途径的激活而抑制了IκB激酶-β(IKKβ)的活性,这反过来又诱导了A549和U251细胞凋亡。IKKβ可以作为藤黄酸的靶标之一。文章中仔细讨论了化合物的制备。笼中的4-氧杂-三环[4.3.1.0 3,7被认为是药效学支架的] dec-2-one蒽酮代表了一种有前途的癌症治疗剂和有用的针对NF-κB途径的探针。DOI:10.1016/j.ejmech.2012.02.029
-
作为产物:描述:2',3',4'-三羟基苯乙酮 在 potassium carbonate 、 magnesium bromide ethyl etherate 、 potassium iodide 作用下, 以 乙醚 、 N,N-二甲基甲酰胺 为溶剂, 反应 18.0h, 生成 3,4-dibenzyloxy-2-hydroxyacetophenone参考文献:名称:基于3,3'-双香豆素的c-Met抑制剂的设计与合成†摘要:基于3,3'-biscoumarin hit 3的优化,合成了基于双香豆素的c-Met抑制剂库,该库从多种香豆素衍生物库中被鉴定为c-Met的非ATP竞争性抑制剂。在这些化合物中,38和40不仅显示出强大的酶活性,IC 50值分别为107 nM和30 nM,而且还抑制了BaF3 / TPR-Met和EBC-1细胞中的c-Met磷酸化。DOI:10.1039/c4ob00364k
文献信息
-
New polyhydroxylated flavon-3-ols and 3-hydroxy-2-styrylchromones: synthesis and ROS/RNS scavenging activities作者:Joana L.C. Sousa、Carina Proença、Marisa Freitas、Eduarda Fernandes、Artur M.S. SilvaDOI:10.1016/j.ejmech.2016.04.057日期:2016.8hydrogen peroxide (H2O2), hypochlorous acid (HOCl), singlet oxygen (1O2) and peroxyl radical (ROO)] and RNS [nitric oxide (NO) and peroxynitrite anion (ONOO−)]. Generally, all the tested new polyhydroxylated flavon-3-ols and 3-hydroxy-2-styrylchromones exhibited scavenging effects dependent on the concentration, and with IC50 values found within the micromolar range. This work allowed the establishment制备了新的多羟基黄酮-3-醇和3-羟基-2-苯乙烯基色酮,并将其评估为活性氧(ROS)和活性氮(RNS)清除剂。合成策略涉及由适当的2'-羟基苯乙酮与苯甲醛/肉桂醛的碱催化醛醇缩合反应制备2'-羟基查耳酮和2'-羟基肉桂基苯乙酮乙酰苯,然后进行Algar-Flynn-Oyamada(AFO)反应以生成聚烷氧基(黄酮3-醇和3-羟基2-苯乙烯基色酮。该合成途径的最后一步包括裂解保护基团,得到预期的多羟基化衍生物。 目前的工作中的研究包括体外对抗生理上最相关的ROS [超氧自由基(O的synthetized化合物的清除活性2 - ),过氧化氢(H 2 ö 2),次氯酸(HOCl),单线态氧(1 Ò 2)和过氧化氢自由基(ROO )]和RNS [一氧化氮(NO)和过氧亚硝酸盐阴离子(ONOO - )。通常,所有测试的新多羟基黄酮-3-醇和3-羟基-2-苯乙烯基色酮均表现出清除作用,该清除作用取决于浓度,并且IC
-
The synthetic flavanone 6-methoxy-2-(naphthalen-1-yl)chroman-4-one induces apoptosis and activation of the MAPK pathway in human U-937 leukaemia cells作者:Ester Saavedra、Henoc Del Rosario、Ignacio Brouard、Judith Hernández-Garcés、Celina García、José Quintana、Francisco EstévezDOI:10.1016/j.bioorg.2019.103450日期:2020.1Synthetic flavonoids containing a naphthalene ring have attracted attention as potential cytotoxic compounds. Here, we synthesized ten chalcones and their corresponding flavanones and evaluated their antiproliferative activity against the human tumour cell line U-937. This series of chalcone derivatives was characterized by the presence of a naphthalene ring which was kept unaltered- and attached to含有萘环的合成类黄酮作为潜在的细胞毒性化合物引起了人们的关注。在这里,我们合成了十个查耳酮及其相应的黄烷酮,并评估了它们对人肿瘤细胞系U-937的抗增殖活性。该系列查尔酮衍生物的特征在于存在萘环,该萘环保持不变并且附着在1-苯基-2-丙烯-1-酮骨架的β碳上。通过引入不同的取代基(甲基,甲氧基,苄氧基,氯)或改变A环上甲氧基或苄氧基的位置,研究了这些查耳酮衍生物及其相应的环状化合物的结构活性关系。结果表明,两个查尔酮均在5'端含有甲氧基 A环位置及其相应的黄烷酮[6-甲氧基-2-(萘-1-基)苯并二氢吡喃-4-酮]是最具细胞毒性的化合物,IC50值为2.8±0.2和1.3±0.2μM,分别针对U-937细胞。这种合成的黄烷酮在U-937细胞中具有与抗肿瘤依托泊苷同等的细胞毒性,并且对其他人类白血病细胞系(包括HL-60,MOLT-3和NALM-6)显示出强大的细胞毒性。人外周血单核细胞比白
-
一种7,8-二羟基黄酮的制备方法
-
Synthesis and Anticancer Activity of 7,8-dihydroxy-4-arylcoumarins作者:Jianrui Wu、Ting Peng、Fang Chen、Yixin Leng、Linjiang Tong、Mengyuan Li、Rong Qu、Hua Xie、Jian Ding、Wenhu DuanDOI:10.2174/1570180812666141111235026日期:2015.3.24A series of 7,8-dihydroxy-4-arylcoumarins, the derivatives related to DW532 that was an anti-tumor agent targeting both kinase and tubulin, was prepared by Suzuki coupling reaction. Among them, compounds 6a, 6b, and 6c were found to exhibit anti-proliferation activities against human breast carcinoma MDA-MB-468 cells with IC50 values of 0.64, 0.69, and 1.33 μM, respectively and human epidermoid carcinoma A431 cells with IC50 values of 2.56, 1.78, and 2.29 μM, respectively. Further evaluation of the selected molecules revealed that they displayed broad-spectrum inhibitory activities against a panel of kinases including Flt-1, VEGFR2, RET, EGFR, etc. In vitro tubulin polymerization assay and molecular docking indicated that the substitution of 3’-OH and 4’-OCH3 on the 4-phenyl ring was essential to achieve potent tubulin inhibition.通过铃木偶联反应制备了一系列7,8-二羟基-4-芳基香豆素,它们是与DW532相关的衍生物,DW532是一种靶向激酶和微管蛋白的抗肿瘤药物。其中,化合物 6a、6b 和 6c 对人乳腺癌 MDA-MB-468 细胞具有抗增殖活性,IC50 值分别为 0.64、0.69 和 1.33 μM;对人表皮样癌 A431 细胞具有抗增殖活性,IC50 值分别为 2.对所选分子的进一步评估表明,它们对包括 Flt-1、血管内皮生长因子受体 2、RET、表皮生长因子受体等在内的一系列激酶具有广谱的抑制活性。体外微管蛋白聚合试验和分子对接表明,4-苯基环上 3'-OH 和 4'-OCH3 的取代是实现强效微管蛋白抑制的关键。
-
Identification of DW532 as a novel anti-tumor agent targeting both kinases and tubulin作者:Ting Peng、Jian-rui Wu、Lin-jiang Tong、Meng-yuan Li、Fang Chen、Yi-xin Leng、Rong Qu、Kun Han、Yi Su、Yi Chen、Wen-hu Duan、Hua Xie、Jian DingDOI:10.1038/aps.2014.33日期:2014.77,8-Dihydroxy-4-(3-hydroxy-4-methoxyphenyl)-2H-chromen-2-one (DW532) is one of simplified analogues of hematoxylin that has shown broad-spectrum inhibition on tyrosine kinases and in vitro anti-cancer activities. The aim of this study was to identify DW532 as a agent targeting both kinases and tubulin, and to investigate its anti-cancer and anti-angiogenesis activities. In vitro tyrosine kinases activity was examined with ELISA, and tyrosine kinases activity in cells was evaluated with Western blot analysis. Tubulin turbidity assay, surface plasmon resonance and immunofluorescence technique were used to characterize the tubulin inhibitory activity. Cell proliferation was examined with SRB assay, and cell apoptosis and cell cycle distribution were analyzed with Annexin-V/PI staining and flow cytometry. Tube formation, aortic ring and chick chorioallantoic membrane assays were used to evaluate the anti-angiogenesis efficacy. DW532 inhibited EGFR and VEGFR2 in vitro kinase activity (the IC50 values were 4.9 and 5.5 μmol/L, respectively), and suppressed their downstream signaling. DW532 dose-dependently inhibited tubulin polymerization via direct binding to tubulin, thus disrupting the mitotic spindle assembly and leading to abnormal cell division. In a panel of human cancer cells, DW532 (1 and 10 μmol/L) induced G2/M phase arrest and cell apoptosis, which subsequently resulted in cytotoxicity. Knockdown of BubR1 or Mps1, the two core proteins of the spindle assembly checkpoint dramatically decreased DW532-induced cell cycle arrest in MDA-MB-468 cells. Moreover, treatment with DW532 potently and dose-dependently suppressed angiogenesis in vitro and in vivo. DW532 is a dual inhibitor against tubulin and tyrosine kinases, and deserves further development as a novel anti-cancer agent.7,8-二羟基-4-(3-羟基-4-甲氧基苯基)-2H-色烯-2-酮(DW532)是一种简化的苏木精类似物,具有广谱抗酪氨酸激酶和体外抗癌活性。本研究旨在确定 DW532 是一种同时针对激酶和微管蛋白的药物,并研究其抗癌和抗血管生成活性。体外酪氨酸激酶活性用酶联免疫吸附试验检测,细胞内酪氨酸激酶活性用 Western 印迹分析评估。利用管蛋白浊度测定法、表面等离子体共振和免疫荧光技术来表征管蛋白的抑制活性。细胞增殖采用 SRB 检测法,细胞凋亡和细胞周期分布采用 Annexin-V/PI 染色法和流式细胞术进行分析。管形成、主动脉环和小鸡绒毛膜试验用于评估抗血管生成的功效。DW532抑制了表皮生长因子受体和血管内皮生长因子受体2的体外激酶活性(IC50值分别为4.9和5.5 μmol/L),并抑制了它们的下游信号转导。DW532 通过与微管蛋白直接结合,剂量依赖性地抑制微管蛋白聚合,从而破坏有丝分裂纺锤体的组装,导致细胞分裂异常。在一组人类癌细胞中,DW532(1 μmol/L 和 10 μmol/L )可诱导 G2/M 期停滞和细胞凋亡,从而产生细胞毒性。敲除纺锤体组装检查点的两个核心蛋白BubR1或Mps1可显著减少DW532诱导的MDA-MB-468细胞的细胞周期停滞。此外,DW532 还能有效抑制体外和体内的血管生成,且其抑制作用与剂量相关。DW532 是一种针对微管蛋白和酪氨酸激酶的双重抑制剂,值得作为一种新型抗癌药物进一步开发。
表征谱图
-
氢谱1HNMR
-
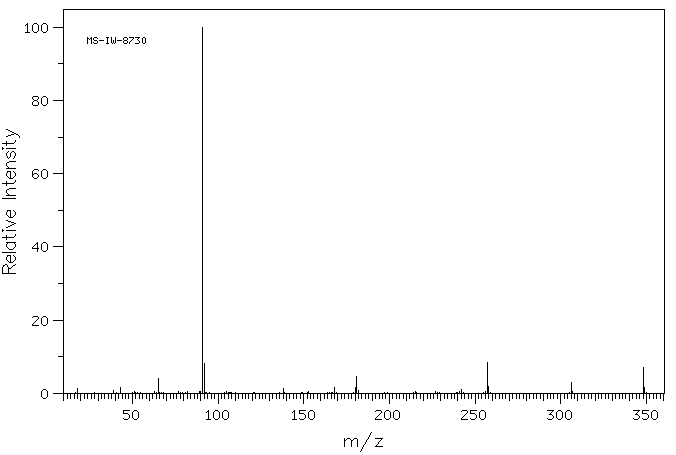
质谱MS
-
碳谱13CNMR
-
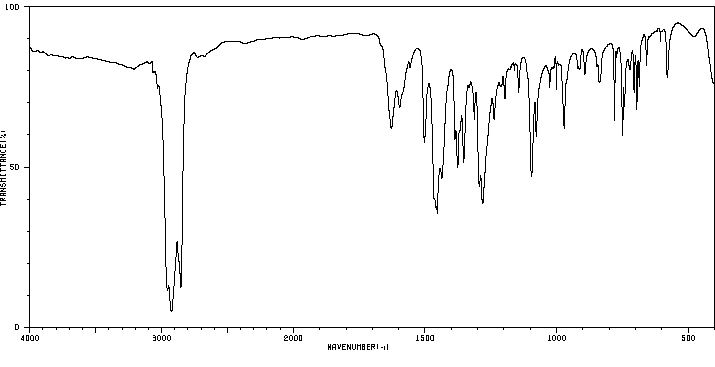
红外IR
-
拉曼Raman
-
峰位数据
-
峰位匹配
-
表征信息
同类化合物
(反式)-4-壬烯醛
(s)-2,3-二羟基丙酸甲酯
([1-(甲氧基甲基)-1H-1,2,4-三唑-5-基](苯基)甲酮)
(Z)-4-辛烯醛
(S)-氨基甲酸酯β-D-O-葡糖醛酸
(S)-3-(((2,2-二氟-1-羟基-7-(甲基磺酰基)-2,3-二氢-1H-茚满-4-基)氧基)-5-氟苄腈
(R)-氨基甲酸酯β-D-O-葡糖醛酸
(5,5-二甲基-2-(哌啶-2-基)环己烷-1,3-二酮)
(2,5-二氟苯基)-4-哌啶基-甲酮
龙胆苦苷
龙胆二糖甲乙酮氰醇(P)
龙胆二糖丙酮氰醇(P)
龙胆三糖
龙涎酮
齐罗硅酮
齐留通beta-D-葡糖苷酸
鼠李糖
黑芥子苷单钾盐
黑海棉酸钠盐
黑木金合欢素
黑曲霉三糖
黑介子苷
黄尿酸8-O-葡糖苷
麻西那霉素II
麦迪霉素
麦芽糖脎
麦芽糖基海藻糖
麦芽糖1-磷酸酯
麦芽糖
麦芽四糖醇
麦芽四糖
麦芽十糖
麦芽六糖
麦芽五糖水合物
麦芽五糖
麦芽五糖
麦芽五糖
麦芽三糖醇
麦芽三糖
麦芽三糖
麦芽三塘水合
麦芽七糖水合物
麦芽七糖
麦法朵
麦可酚酸-酰基-Β-D-葡糖苷酸
麦利查咪
麝香酮
鹤草酚
鸢尾酚酮 3-C-beta-D-吡喃葡萄糖苷
鸡矢藤苷

联系我们



关注我们

公众号

