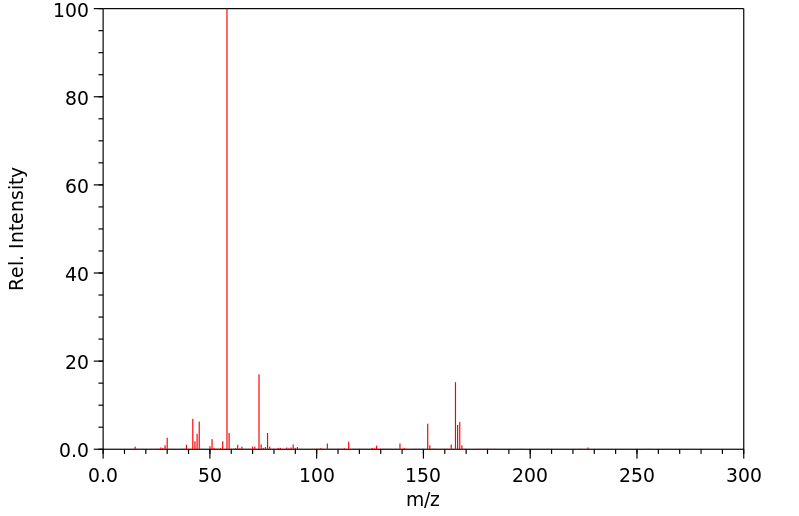
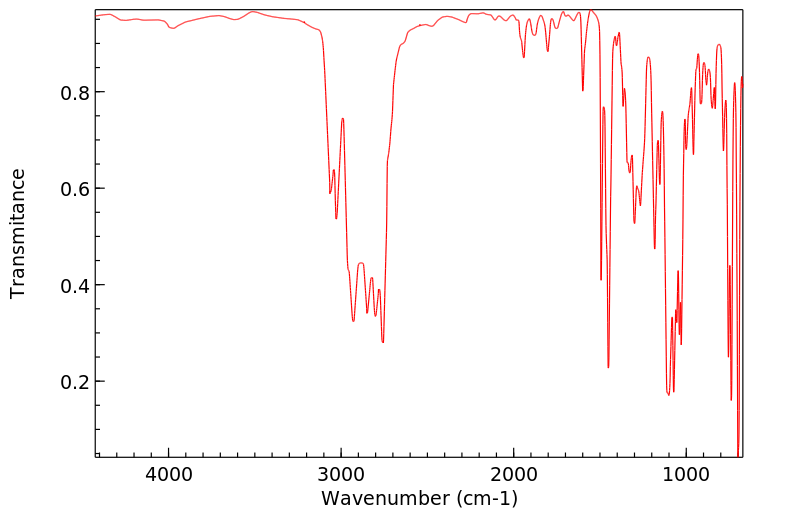
Diphenhydramine undergoes rapid and extensive first-pass metabolism. In particular, two successive N-demethylations occur wherein diphenhydramine is demethylated to N-desmethyldiphenhydramine (the N-desmethyl metabolite) and then this metabolite is itself demethylated to N,N-didesmethyldiphenhydramine (the N,N-didesmethyl metabolite). Subsequently, acetyl metabolites like N-acetyl-N-desmethyldiphenhydramine are generated via the amine moiety of the N,N-didesmethyl metabolite. Additionally, the N,N-didesmethyl metabolite also undergoes some oxidation to generate the diphenylmethoxyacetic acid metabolite as well. The remaining percentage of a dose of administered diphenhydramine is excreted unchanged. The metabolites are further conjugated with glycine and glutamine and excreted in urine. Moreover, studies have determined that a variety of cytochrome P450 isoenzymes are involved in the N-demethylation that characterizes the primary metabolic pathway of diphenhydramine, including CYP2D6, CYP1A2, CYP2C9, and CYP2C19. In particular, CYP2D6 demonstrates higher affinity catalysis with the diphenhydramine substrate than the other isoenzymes identified. Consequently, inducers or inhibitors of these such CYP enzymes may potentially affect the serum concentration and incidence and/or severity of adverse effects associated with exposure to diphenhydramine.
Diphenhydramine is rapidly and apparently almost completely metabolized. Following oral administration, the drug apparently undergoes substantial first-pass metabolism in the liver. Diphenhydramine appears to be metabolized principally to diphenylmethoxyacetic acid, which may further undergo conjugation. The drug also undergoes dealkylation to form the N-demethyl and N, N-didemethyl derivatives. Diphenhydramine and its metabolites are excreted principally in urine.
Diphenhydramine is widely used as an over-the-counter antihistamine. However, the specific human cytochrome P450 (P450) isozymes that mediate the metabolism of diphenhydramine in the range of clinically relevant concentrations (0.14-0.77 microM) remain unclear. Therefore, P450 isozymes involved in N-demethylation, a main metabolic pathway of diphenhydramine, were identified by a liquid chromatography-mass spectrometry method developed in our laboratory. Among 14 recombinant P450 isozymes, CYP2D6 showed the highest activity of diphenhydramine N-demethylation (0.69 pmol/min/pmol P450) at 0.5 uM. CYP2D6 catalyzed diphenhydramine N-demethylation as a high-affinity P450 isozyme, the K(m) value of which was 1.12 +/- 0.21 uM. In addition, CYP1A2, CYP2C9, and CYP2C19 were identified as low-affinity components. In human liver microsomes, involvement of CYP2D6, CYP1A2, CYP2C9, and CYP2C19 in diphenhydramine N-demethylation was confirmed by using P450 isozyme-specific inhibitors. In addition, contributions of these P450 isozymes estimated by the relative activity factor were in good agreement with the results of inhibition studies. Although an inhibitory effect of diphenhydramine on the metabolic activity of CYP2D6 has been reported previously, the results of the present study suggest that it is not only a potent inhibitor but also a high-affinity substrate of CYP2D6. Therefore, it is worth mentioning that the sedative effect of diphenhydramine might be caused by coadministration of CYP2D6 substrate(s)/inhibitor(s). In addition, large differences in the metabolic activities of CYP2D6 and those of CYP1A2, CYP2C9, and CYP2C19 could cause the individual differences in anti-allergic efficacy and the sedative effect of diphenhydramine.
Two strains of the filamentous fungus Cunninghamella elegans (ATCC 9245 and ATCC 36112) were grown in Sabouraud dextrose broth and screened for the ability to metabolize the ethanolamine-type antihistamine diphenhydramine. Based on the amount of parent drug recovered after 7 days incubation, both C. elegans strains metabolized approximately 74% of the diphenhydramine, 58% of this being identified as organic extractable metabolites. The organic extractable metabolites were isolated by reversed-phase high-performance liquid chromatography and identified by analyzing their mass and nuclear magnetic resonance spectra. Desorption chemical ionization mass spectrometry (DCIMS) with deuterated ammonia was used to differentiate possible isobaric diphenhydramine metabolites and to probe the mechanisms of ion formation under ammonia DCIMS conditions. C. elegans transformed diphenhydramine by demethylation, oxidation, and N-acetylation. The major metabolites observed were diphenhydramine-N-oxide (3%), N-desmethyldiphenhydramine (30%), N-acetyldidesmethyldiphenhydramine (13%), and N-acetyl-N-desmethyldiphenhydramine (12%). These compounds are known mammalian metabolites of diphenhydramine ... .
Diphenhydramine competes with free histamine for binding at HA-receptor sites. This antagonizes the effects of histamine on HA-receptors, leading to a reduction of the negative symptoms brought on by histamine HA-receptor binding.
Despite widespread use over many decades, diphenhydramine has not been linked to liver test abnormalities or to clinically apparent liver injury. The reason for its safety may relate its short half-life and limited duration of use.
Likelihood score: E (unlikely to be a cause of clinically apparent liver injury).
References on the safety and potential hepatotoxicity of antihistamines are given together after the Overview section on Antihistamines.
Drug Class: Antihistamines
Diphenhydramine is quickly absorbed after oral administration with maximum activity occurring in approximately one hour. The oral bioavailability of diphenhydramine has been documented in the range of 40% to 60%, and peak plasma concentration occurs about 2 to 3 hours after administration.
The metabolites of diphenhydramine are conjugated with glycine and glutamine and excreted in urine. Only about 1% of a single dose is excreted unchanged in urine. The medication is ultimately eliminated by the kidneys slowly, mainly as inactive metabolites.
Diphenhydramine is widely distributed throughout the body, including the CNS. Following a 50 mg oral dose of diphenhydramine, the volume of distribution is in the range of 3.3 - 6.8 l/kg.
Distribution of diphenhydramine into human body tissues and fluids has not been fully characterized. Following IV administration in rats, highest concentrations of the drug are attained in the lungs, spleen, and brain, with lower concentrations in the heart, muscle, and liver. Following IV administration in healthy adults, diphenhydramine reportedly has an apparent volume of distribution of 188-336 L. Volume of distribution of the drug reportedly is larger in Asian (about 480 L) than white adults.
The invention relates to novel 3-amino pyrrolidine derivatives, as well as methods for modulating calcium channel activity and for treating conditions associated with calcium channel function. In particular, the compounds generally contain at least one benzhydril moiety, and are useful in treating conditions which benefit from blocking calcium ion channels.
Compositions for Treatment of Cystic Fibrosis and Other Chronic Diseases
申请人:Vertex Pharmaceuticals Incorporated
公开号:US20150231142A1
公开(公告)日:2015-08-20
The present invention relates to pharmaceutical compositions comprising an inhibitor of epithelial sodium channel activity in combination with at least one ABC Transporter modulator compound of Formula A, Formula B, Formula C, or Formula D. The invention also relates to pharmaceutical formulations thereof, and to methods of using such compositions in the treatment of CFTR mediated diseases, particularly cystic fibrosis using the pharmaceutical combination compositions.
[EN] SUBSTITUTED 4-PYRIDONES AND THEIR USE AS INHIBITORS OF NEUTROPHIL ELASTASE ACTIVITY<br/>[FR] 4-PYRIDONES SUBSTITUÉES ET LEUR UTILISATION COMME INHIBITEURS DE L'ACTIVITÉ DE L'ÉLASTASE NEUTROPHILE
申请人:BOEHRINGER INGELHEIM INT
公开号:WO2014029831A1
公开(公告)日:2014-02-27
This invention relates to substituted 4-pyridones of formula 1 and their use as inhibitors of neutrophil elastase activity, pharmaceutical compositions containing the same, and methods of using the same as agents for treatment and/or prevention of pulmonary, gastrointestinal and genitourinary diseases, inflammatory diseases of the skin and the eye and other auto-immune and allergic disorders, allograft rejection, and oncological diseases.
SUBSTITUTED 4-PYRIDONES AND THEIR USE AS INHIBITORS OF NEUTROPHIL ELASTASE ACTIVITY
申请人:OOST Thorsten
公开号:US20140057916A1
公开(公告)日:2014-02-27
This invention relates to substituted 4-pyridones of formula 1
and their use as inhibitors of neutrophil elastase activity, pharmaceutical compositions containing the same, and methods of using the same as agents for treatment and/or prevention of pulmonary, gastrointestinal and genitourinary diseases, inflammatory diseases of the skin and the eye and other auto-immune and allergic disorders, allograft rejection, and oncological diseases.
[EN] NOVEL COMPOUNDS AND PHARMACEUTICAL COMPOSITIONS THEREOF FOR THE TREATMENT OF INFLAMMATORY DISORDERS<br/>[FR] NOUVEAUX COMPOSÉS ET COMPOSITIONS PHARMACEUTIQUES LES COMPRENANT POUR LE TRAITEMENT DE TROUBLES INFLAMMATOIRES
申请人:GALAPAGOS NV
公开号:WO2017012647A1
公开(公告)日:2017-01-26
The present invention discloses compounds according to Formula (I), wherein R1, R3, R4, R5, L1, and Cy are as defined herein. The present invention also provides compounds, methods for the production of said compounds of the invention, pharmaceutical compositions comprising the same and their use in allergic or inflammatory conditions, autoimmune diseases, proliferative diseases, transplantation rejection, diseases involving impairment of cartilage turnover, congenital cartilage malformations, and/or diseases associated with hypersecretion of IL6 and/or interferons. The present invention also methods for the prevention and/or treatment of the aforementioned diseases by administering a compound of the invention.