
2-(2-氨乙基)-1,3-二氧戊环 | 5754-35-8
中文名称
2-(2-氨乙基)-1,3-二氧戊环
中文别名
2-(2-氨基乙基)-1,3-二氧戊烷
英文名称
2-[1,3]dioxolan-2-yl-ethylamine
英文别名
2-(2-aminoethyl)-1,3-dioxolane;2-(1,3-dioxolan-2-yl)ethanamine;2-(aminoethyl)-1,3-dioxolane

CAS
5754-35-8
化学式
C5H11NO2
mdl
MFCD00142756
分子量
117.148
InChiKey
JNAQYWWCTUEVKR-UHFFFAOYSA-N
BEILSTEIN
——
EINECS
——
-
物化性质
-
计算性质
-
ADMET
-
安全信息
-
SDS
-
制备方法与用途
-
上下游信息
-
文献信息
-
表征谱图
-
同类化合物
-
相关功能分类
-
相关结构分类
物化性质
-
沸点:75 °C / 18mmHg
-
密度:1,07 g/cm3
-
溶解度:可溶于二氯甲烷、甲醇
-
稳定性/保质期:
在常温常压下保持稳定
计算性质
-
辛醇/水分配系数(LogP):-0.7
-
重原子数:8
-
可旋转键数:2
-
环数:1.0
-
sp3杂化的碳原子比例:1.0
-
拓扑面积:44.5
-
氢给体数:1
-
氢受体数:3
安全信息
-
危险等级:3/8
-
安全说明:S16,S26,S36/37/39
-
危险类别码:R36/37/38,R11
-
海关编码:2932999099
-
危险品运输编号:1993
-
储存条件:常温下,应避光存放于阴凉干燥处,并密封保存。
SDS
2-(2-氨乙基)-1,3-二氧戊环 修改号码:5
模块 1. 化学品
产品名称: 2-(2-Aminoethyl)-1,3-dioxolane
修改号码: 5
模块 2. 危险性概述
GHS分类
物理性危害
易燃液体 第3级
健康危害
皮肤腐蚀/刺激 1C类
严重损伤/刺激眼睛 第1级
环境危害 未分类
GHS标签元素
图标或危害标志
信号词 危险
危险描述 易燃液体和蒸气
造成严重的皮肤灼伤和眼损伤
防范说明
[预防] 远离热源/火花/明火/热表面。禁烟。
保持容器密闭。
使用防爆的电气/通风/照明设备。采取预防措施以防静电和火花引起的着火。
切勿吸入。
处理后要彻底清洗双手。
穿戴防护手套/护目镜/防护面具。
[急救措施] 吸入:将受害者移到新鲜空气处,在呼吸舒适的地方保持休息。
食入:漱口。切勿催吐。
眼睛接触:用水小心清洗几分钟。如果方便,易操作,摘除隐形眼镜。继续冲洗。
皮肤接触:立即去除/脱掉所有被污染的衣物。用水清洗皮肤/淋浴。
被污染的衣物清洗后方可重新使用。
立即呼叫解毒中心/医生。
[储存] 存放于通风良好处。保持凉爽。
存放处须加锁。
2-(2-氨乙基)-1,3-二氧戊环 修改号码:5
模块 2. 危险性概述
[废弃处置] 根据当地政府规定把物品/容器交与工业废弃处理机构。
模块 3. 成分/组成信息
单一物质/混和物 单一物质
化学名(中文名): 2-(2-氨乙基)-1,3-二氧戊环
百分比: >98.0%(GC)(T)
CAS编码: 5754-35-8
分子式:
C5H11NO2
模块 4. 急救措施
吸入: 将受害者移到新鲜空气处,保持呼吸通畅,休息。立即呼叫解毒中心/医生。
皮肤接触: 立即去除/脱掉所有被污染的衣物。用大量肥皂和水轻轻洗。
立即呼叫解毒中心/医生。
眼睛接触: 用水小心清洗几分钟。如果方便,易操作,摘除隐形眼镜。继续清洗。
立即呼叫解毒中心/医生。
食入: 立即呼叫解毒中心/医生。漱口。切勿引吐。
紧急救助者的防护: 救援者需要穿戴个人防护用品,比如橡胶手套和气密性护目镜。
模块 5. 消防措施
合适的灭火剂: 干粉,泡沫,雾状水,二氧化碳
不适用的灭火剂: 棒状水
特殊危险性: 小心,燃烧或高温下可能分解产生毒烟。
特定方法: 从上风处灭火,根据周围环境选择合适的灭火方法。
非相关人员应该撤离至安全地方。
周围一旦着火:喷水,保持容器冷却。如果安全,消除一切火源。
消防员的特殊防护用具: 灭火时,一定要穿戴个人防护用品。
模块 6. 泄漏应急处理
个人防护措施,防护用具, 使用特殊的个人防护用品(自携式呼吸器)。远离溢出物/泄露处并处在上风处。确保
紧急措施: 足够通风。
泄露区应该用安全带等圈起来,控制非相关人员进入。
环保措施: 防止进入下水道。
控制和清洗的方法和材料: 回收到密闭容器前用干砂或惰性吸收剂吸收泄漏物。一旦大量泄漏,筑堤控制。附着
物或收集物应该根据相关法律法规废弃处置。
副危险性的防护措施 移除所有火源。一旦发生火灾应该准备灭火器。使用防火花工具和防爆设备。
模块 7. 操作处置与储存
处理
技术措施: 在通风良好处进行处理。穿戴合适的防护用具。防止烟雾产生。远离热源/火花/明火
/热表面。禁烟。采取措施防止静电积累。使用防爆设备。处理后彻底清洗双手和脸。
注意事项: 如果可能,使用封闭系统。如果蒸气或浮质产生,使用通风、局部排气。
操作处置注意事项: 避免接触皮肤、眼睛和衣物。
贮存
储存条件: 保持容器密闭。存放于冷藏防爆冰箱。
存放于惰性气体环境中。
防湿。
存放处须加锁。
远离不相容的材料比如氧化剂存放。
热敏, 易湿
2-(2-氨乙基)-1,3-二氧戊环 修改号码:5
模块 7. 操作处置与储存
包装材料: 依据法律。
模块 8. 接触控制和个体防护
工程控制: 尽可能安装封闭体系或局部排风系统。同时安装淋浴器和洗眼器。
个人防护用品
呼吸系统防护: 半面罩或全面罩呼吸器,自携式呼吸器(SCBA),供气呼吸器等。依据当地和政府法
规,使用通过政府标准的呼吸器。
手部防护: 防渗手套。
眼睛防护: 护目镜。如果情况需要,佩戴面具。
皮肤和身体防护: 防渗防护服。如果情况需要,穿戴防护靴。
模块 9. 理化特性
液体
外形(20°C):
外观: 透明
颜色: 无色-几乎无色
气味: 无资料
pH: 无数据资料
熔点: 无资料
沸点/沸程 75 °C/2.4kPa
闪点: 无资料
爆炸特性
爆炸下限: 无资料
爆炸上限: 无资料
密度: 1.07
溶解度:
[水] 无资料
[其他溶剂] 无资料
模块 10. 稳定性和反应性
化学稳定性: 一般情况下稳定。
危险反应的可能性: 未报道特殊反应性。
避免接触的条件: 火花, 明火, 静电
须避免接触的物质 氧化剂
危险的分解产物: 一氧化碳, 二氧化碳, 氮氧化物 (NOx)
模块 11. 毒理学信息
急性毒性: 无资料
对皮肤腐蚀或刺激: 无资料
对眼睛严重损害或刺激: 无资料
生殖细胞变异原性: 无资料
致癌性:
IARC = 无资料
NTP = 无资料
生殖毒性: 无资料
模块 12. 生态学信息
生态毒性:
鱼类: 无资料
甲壳类: 无资料
2-(2-氨乙基)-1,3-二氧戊环 修改号码:5
模块 12. 生态学信息
藻类: 无资料
残留性 / 降解性: 无资料
潜在生物累积 (BCF): 无资料
土壤中移动性
log水分配系数: 无资料
土壤吸收系数 (Koc): 无资料
亨利定律 无资料
constaNT(PaM3/mol):
模块 13. 废弃处置
如果可能,回收处理。请咨询当地管理部门。建议在装有后燃和洗涤装置的化学焚烧炉中焚烧。废弃处置时请遵守
国家、地区和当地的所有法规。
模块 14. 运输信息
联合国分类: 第3类 易燃液体 。
副危险性: 第8类 腐蚀品
UN编号: 2733
正式运输名称: 胺类, 易燃的, 腐蚀的, 不另作详细说明
包装等级: III
模块 15. 法规信息
《危险化学品安全管理条例》(2002年1月26日国务院发布,2011年2月16日修订): 针对危险化学品的安全使用、
生产、储存、运输、装卸等方面均作了相应的规定。
模块16 - 其他信息
N/A
模块 1. 化学品
产品名称: 2-(2-Aminoethyl)-1,3-dioxolane
修改号码: 5
模块 2. 危险性概述
GHS分类
物理性危害
易燃液体 第3级
健康危害
皮肤腐蚀/刺激 1C类
严重损伤/刺激眼睛 第1级
环境危害 未分类
GHS标签元素
图标或危害标志
信号词 危险
危险描述 易燃液体和蒸气
造成严重的皮肤灼伤和眼损伤
防范说明
[预防] 远离热源/火花/明火/热表面。禁烟。
保持容器密闭。
使用防爆的电气/通风/照明设备。采取预防措施以防静电和火花引起的着火。
切勿吸入。
处理后要彻底清洗双手。
穿戴防护手套/护目镜/防护面具。
[急救措施] 吸入:将受害者移到新鲜空气处,在呼吸舒适的地方保持休息。
食入:漱口。切勿催吐。
眼睛接触:用水小心清洗几分钟。如果方便,易操作,摘除隐形眼镜。继续冲洗。
皮肤接触:立即去除/脱掉所有被污染的衣物。用水清洗皮肤/淋浴。
被污染的衣物清洗后方可重新使用。
立即呼叫解毒中心/医生。
[储存] 存放于通风良好处。保持凉爽。
存放处须加锁。
2-(2-氨乙基)-1,3-二氧戊环 修改号码:5
模块 2. 危险性概述
[废弃处置] 根据当地政府规定把物品/容器交与工业废弃处理机构。
模块 3. 成分/组成信息
单一物质/混和物 单一物质
化学名(中文名): 2-(2-氨乙基)-1,3-二氧戊环
百分比: >98.0%(GC)(T)
CAS编码: 5754-35-8
分子式:
C5H11NO2
模块 4. 急救措施
吸入: 将受害者移到新鲜空气处,保持呼吸通畅,休息。立即呼叫解毒中心/医生。
皮肤接触: 立即去除/脱掉所有被污染的衣物。用大量肥皂和水轻轻洗。
立即呼叫解毒中心/医生。
眼睛接触: 用水小心清洗几分钟。如果方便,易操作,摘除隐形眼镜。继续清洗。
立即呼叫解毒中心/医生。
食入: 立即呼叫解毒中心/医生。漱口。切勿引吐。
紧急救助者的防护: 救援者需要穿戴个人防护用品,比如橡胶手套和气密性护目镜。
模块 5. 消防措施
合适的灭火剂: 干粉,泡沫,雾状水,二氧化碳
不适用的灭火剂: 棒状水
特殊危险性: 小心,燃烧或高温下可能分解产生毒烟。
特定方法: 从上风处灭火,根据周围环境选择合适的灭火方法。
非相关人员应该撤离至安全地方。
周围一旦着火:喷水,保持容器冷却。如果安全,消除一切火源。
消防员的特殊防护用具: 灭火时,一定要穿戴个人防护用品。
模块 6. 泄漏应急处理
个人防护措施,防护用具, 使用特殊的个人防护用品(自携式呼吸器)。远离溢出物/泄露处并处在上风处。确保
紧急措施: 足够通风。
泄露区应该用安全带等圈起来,控制非相关人员进入。
环保措施: 防止进入下水道。
控制和清洗的方法和材料: 回收到密闭容器前用干砂或惰性吸收剂吸收泄漏物。一旦大量泄漏,筑堤控制。附着
物或收集物应该根据相关法律法规废弃处置。
副危险性的防护措施 移除所有火源。一旦发生火灾应该准备灭火器。使用防火花工具和防爆设备。
模块 7. 操作处置与储存
处理
技术措施: 在通风良好处进行处理。穿戴合适的防护用具。防止烟雾产生。远离热源/火花/明火
/热表面。禁烟。采取措施防止静电积累。使用防爆设备。处理后彻底清洗双手和脸。
注意事项: 如果可能,使用封闭系统。如果蒸气或浮质产生,使用通风、局部排气。
操作处置注意事项: 避免接触皮肤、眼睛和衣物。
贮存
储存条件: 保持容器密闭。存放于冷藏防爆冰箱。
存放于惰性气体环境中。
防湿。
存放处须加锁。
远离不相容的材料比如氧化剂存放。
热敏, 易湿
2-(2-氨乙基)-1,3-二氧戊环 修改号码:5
模块 7. 操作处置与储存
包装材料: 依据法律。
模块 8. 接触控制和个体防护
工程控制: 尽可能安装封闭体系或局部排风系统。同时安装淋浴器和洗眼器。
个人防护用品
呼吸系统防护: 半面罩或全面罩呼吸器,自携式呼吸器(SCBA),供气呼吸器等。依据当地和政府法
规,使用通过政府标准的呼吸器。
手部防护: 防渗手套。
眼睛防护: 护目镜。如果情况需要,佩戴面具。
皮肤和身体防护: 防渗防护服。如果情况需要,穿戴防护靴。
模块 9. 理化特性
液体
外形(20°C):
外观: 透明
颜色: 无色-几乎无色
气味: 无资料
pH: 无数据资料
熔点: 无资料
沸点/沸程 75 °C/2.4kPa
闪点: 无资料
爆炸特性
爆炸下限: 无资料
爆炸上限: 无资料
密度: 1.07
溶解度:
[水] 无资料
[其他溶剂] 无资料
模块 10. 稳定性和反应性
化学稳定性: 一般情况下稳定。
危险反应的可能性: 未报道特殊反应性。
避免接触的条件: 火花, 明火, 静电
须避免接触的物质 氧化剂
危险的分解产物: 一氧化碳, 二氧化碳, 氮氧化物 (NOx)
模块 11. 毒理学信息
急性毒性: 无资料
对皮肤腐蚀或刺激: 无资料
对眼睛严重损害或刺激: 无资料
生殖细胞变异原性: 无资料
致癌性:
IARC = 无资料
NTP = 无资料
生殖毒性: 无资料
模块 12. 生态学信息
生态毒性:
鱼类: 无资料
甲壳类: 无资料
2-(2-氨乙基)-1,3-二氧戊环 修改号码:5
模块 12. 生态学信息
藻类: 无资料
残留性 / 降解性: 无资料
潜在生物累积 (BCF): 无资料
土壤中移动性
log水分配系数: 无资料
土壤吸收系数 (Koc): 无资料
亨利定律 无资料
constaNT(PaM3/mol):
模块 13. 废弃处置
如果可能,回收处理。请咨询当地管理部门。建议在装有后燃和洗涤装置的化学焚烧炉中焚烧。废弃处置时请遵守
国家、地区和当地的所有法规。
模块 14. 运输信息
联合国分类: 第3类 易燃液体 。
副危险性: 第8类 腐蚀品
UN编号: 2733
正式运输名称: 胺类, 易燃的, 腐蚀的, 不另作详细说明
包装等级: III
模块 15. 法规信息
《危险化学品安全管理条例》(2002年1月26日国务院发布,2011年2月16日修订): 针对危险化学品的安全使用、
生产、储存、运输、装卸等方面均作了相应的规定。
模块16 - 其他信息
N/A
上下游信息
-
上游原料
中文名称 英文名称 CAS号 化学式 分子量 2-(2-叠氮基乙基)-1,3-二氧戊环 2-(2-azidoethyl)-1,3-dioxolane 111752-08-0 C5H9N3O2 143.145 2-(2-硝基乙基)-[1,3]二氧戊烷 2-(2-nitroethyl)-1,3-dioxolane 82891-99-4 C5H9NO4 147.131 2-(2-溴乙基)-1,3-二恶烷 2-bromo-2-(2-ethyl)dioxolane 18742-02-4 C5H9BrO2 181.029 -
下游产品
中文名称 英文名称 CAS号 化学式 分子量 —— 1-[2-(1,3-dioxolan-2-yl)ethyl]thiourea 957142-30-2 C6H12N2O2S 176.239
反应信息
-
作为反应物:描述:2-(2-氨乙基)-1,3-二氧戊环 在 溶剂黄146 、 zinc(II) chloride 作用下, 以 乙醇 、 甲苯 为溶剂, 反应 50.0h, 生成 1-(2-[1,3]Dioxolan-2-yl-ethyl)-2-methyl-4,5-diphenyl-1H-pyrrole-3-carboxylic acid ethyl ester参考文献:名称:Inhibitors of cholesterol biosynthesis. 3. Tetrahydro-4-hydroxy-6-[2-(1H-pyrrol-1-yl)ethyl]-2H-pyran 2-one inhibitors of HMG-CoA reductase. 2. Effects of introducing substituents at positions three and four of the pyrrole nucleus摘要:A series of trans-tetrahydro-4-hydroxy-6-[2-(2,3,4,5-substituted-1H-pyrrol-1-yl)ethyl]-2H-pyran-2-ones and their dihydroxy acids were prepared and tested for their ability to inhibit the enzyme HMG-CoA reductase in vitro. Inhibitory potency was found to increase substantially when substituents were introduced into positions three and four of the pyrrole ring. A systematic exploration of structure-activity relationships at these two positions led to the identification of a compound ((+)-33, (+)-(4R)-trans-2-(4-fluorophenyl)-5-(1-methylethyl)-N,3-diphenyl-1-[(tetrahydro-4-hydroxy-6-oxo-2H-pyran-2-yl)ethyl]-1H-pyrrole-4-carboxamide) with five times the inhibitory potency of the fungal metabolite compactin.DOI:10.1021/jm00105a056
-
作为产物:描述:2-溴甲基-1,3-二氧戊烷 在 三甲基氰硅烷 、 四丁基氟化铵 、 aluminum (III) chloride 、 lithium aluminium tetrahydride 作用下, 以 乙腈 、 乙醚 为溶剂, 生成 2-(2-氨乙基)-1,3-二氧戊环参考文献:名称:[EN] CLEAVABLE NUCLEOTIDE ANALOGS AND USES THEREOF
[FR] ANALOGUES NUCLÉOTIDIQUES CLIVABLES ET LEURS UTILISATIONS摘要:可切割的核苷酸类似物已提供。该核苷酸类似物包括连接到可切割基团的核苷酸分子,其中可切割基团包括保护基团和/或连接到荧光团的连接物。可切割基团连接到核苷酸分子的五碳糖的3'-OH的氧原子。这些核苷酸类似物可用于使用模板无关聚合酶制备多核苷酸分子。这些核苷酸类似物在DNA合成测序过程中可作为可逆终止子。切割可切割基团恢复自由的3'-OH官能团,从而促进多核苷酸分子的生长。还提供了核苷酸类似物的一般结构以及拟议的合成方案。公开号:WO2018102554A1 -
作为试剂:描述:3-fluoro-N,N-bis(3-methylbutyl)-4-nitrobenzamide 在 2-(2-氨乙基)-1,3-二氧戊环 作用下, 以 乙腈 为溶剂, 反应 3.0h, 以98%的产率得到3-{[2-(1,3-dioxolan-2-yl)ethyl]amino}-N,N-bis(3-methylbutyl)-4-nitrobenzamide参考文献:名称:Novel derivatives of benzimidazole and imidazo-pyridine and their use as medicaments摘要:具有以下结构的化合物及其药用盐具有良好的亲和力,适用于治疗包括疼痛、炎症等在内的疾病,其中取代基如规范中所定义的亚型黑素皮质激素受体。公开号:US20050065179A1
文献信息
-
Chan–Evans–Lam Amination of Boronic Acid Pinacol (BPin) Esters: Overcoming the Aryl Amine Problem作者:Julien C. Vantourout、Robert P. Law、Albert Isidro-Llobet、Stephen J. Atkinson、Allan J. B. WatsonDOI:10.1021/acs.joc.6b00466日期:2016.5.6The Chan–Evans–Lam reaction is a valuable C–N bond forming process. However, aryl boronic acid pinacol (BPin) ester reagents can be difficult coupling partners that often deliver low yields, in particular in reactions with aryl amines. Herein, we report effective reaction conditions for the Chan–Evans–Lam amination of aryl BPin with alkyl and aryl amines. A mixed MeCN/EtOH solvent system was found
-
Radical cyclization in heterocycle synthesis. Part 13: Sulfanyl radical addition–cyclization of oxime ethers and hydrazones connected with alkenes for synthesis of cyclic β-amino acids作者:Okiko Miyata、Kanami Muroya、Tomoko Kobayashi、Rina Yamanaka、Seiko Kajisa、Junko Koide、Takeaki NaitoDOI:10.1016/s0040-4020(02)00412-x日期:2002.5combination of sulfanyl radical addition–cyclization of the oxime ethers and hydrazones connected with alkenes and subsequent conversion of a phenylsulfanylmethyl group to a carboxyl group provides a novel method for the construction of the cyclic β-amino acids. Upon treatment with thiophenol in the presence of AIBN, the oxime ethers and hydrazones smoothly underwent sulfanyl radical addition-cyclization
-
CuI/Oxalic Diamide Catalyzed Coupling Reaction of (Hetero)Aryl Chlorides and Amines作者:Wei Zhou、Mengyang Fan、Junli Yin、Yongwen Jiang、Dawei MaDOI:10.1021/jacs.5b08411日期:2015.9.23A class of oxalic diamides are found to be effective ligands for promoting CuI-catalyzed aryl amination with less reactive (hetero)aryl chlorides. The reaction proceeds at 120 °C with K3PO4 as the base in DMSO to afford a wide range of (hetero)aryl amines in good to excellent yields. The bis(N-aryl) substituted oxalamides are superior ligands to N-aryl-N'-alkyl substituted or bis(N-alkyl) substituted
-
In Situ Proteome Profiling and Bioimaging Applications of Small-Molecule Affinity-Based Probes Derived From DOT1L Inhibitors作者:Biwei Zhu、Hailong Zhang、Sijun Pan、Chenyu Wang、Jingyan Ge、Jun-Seok Lee、Shao Q. YaoDOI:10.1002/chem.201600259日期:2016.6.1which were cell‐permeable and structural mimics of FED1. The binding and inhibitory effects of these two probes against DOT1L protein were extensively investigated in vitro and in live mammalian cells (in situ). The cellular uptake and sub‐cellular localization properties of the probes were subsequently studied in live‐cell imaging experiments, and our results revealed that, whereas both P1 and P2 readilyDOT1L是使赖氨酸79(H3K79)上的组蛋白H3甲基化的唯一蛋白质甲基转移酶,并且是一种有前途的抗癌药物。DOT1L的小分子抑制剂(例如FED1)是潜在的抗癌药,是研究DOT1L在人类疾病中的生物学作用的有用工具。FED1对DOT1L表现出优异的体外抑制活性,但其细胞作用相对较差。在这项研究中,我们设计并合成了具有光反应性和“可点击”亲和力的探针(AfBPs)P1和P2,它们是FED1的细胞渗透性和结构类似物。在体外和活的哺乳动物细胞(原位)中广泛研究了这两种探针对DOT1L蛋白的结合和抑制作用。随后在活细胞成像实验中研究了探针的细胞摄取和亚细胞定位特性,我们的结果表明,尽管P1和P2都容易进入哺乳动物细胞,但大多数不能到达细胞核,功能DOT1L驻留。这为FED1的细胞活性差提供了一个合理的解释。最后,使用P1 / P2进行大规模基于细胞的蛋白质组分析,然后进行定量LC-MS / MS
-
[EN] THIAZOLECARBOXAMIDE DERIVATIVES FOR USE AS NAMPT INHIBITORS<br/>[FR] DÉRIVÉS DE THIAZOLECARBOXAMIDE UTILES EN TANT QU'INHIBITEURS DE LA NAMPT
表征谱图
-
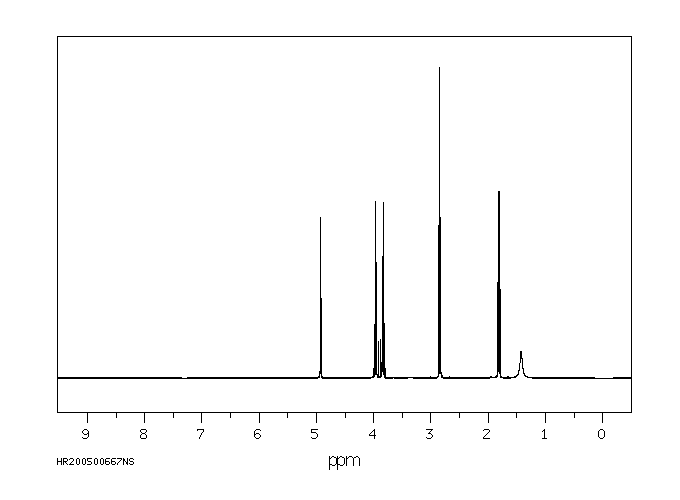
氢谱1HNMR
-
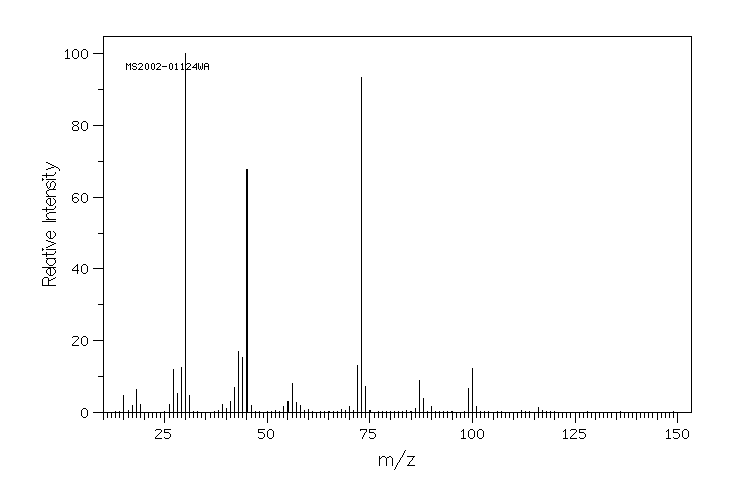
质谱MS
-
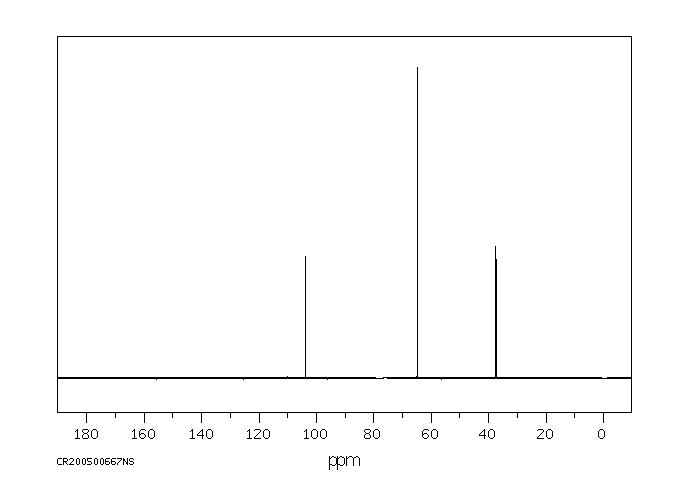
碳谱13CNMR
-
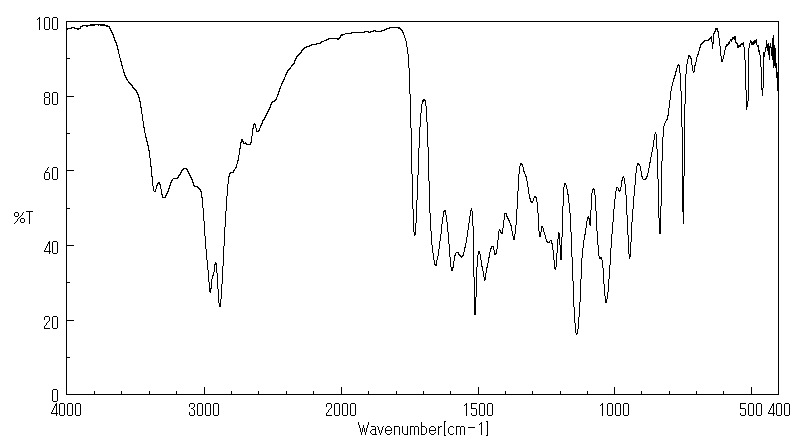
红外IR
-
拉曼Raman
-
峰位数据
-
峰位匹配
-
表征信息
同类化合物
顺式-2-甲基-4-叔-丁基-1,3-二氧戊环
过氧竹红菌素
辛醛丙二醇缩醛
碘丙甘油
甜瓜醛丙二醇缩醛
甘油缩甲醛
甘油缩甲醛
环辛基甲醛乙烯缩醛
环戊二烯内过氧化物
环己丙胺,1-(1,3-二噁戊环-2-基)-
环丙羧酸,2-乙酰基-,甲基酯,(1R-顺)-(9CI)
氯乙醛缩乙二醇
柠檬醛乙二醇缩醛
异戊醛丙二醇缩醛
异丁醛-丙二醇缩醛
奥普碘铵
多米奥醇
多效缩醛
壬醛丙二醇缩醛
四吖戊啶,5-(1-吡咯烷基)-
亲和素
二氰苯乙烯酮乙烯缩醛
乙酮,1-(2-环辛烯-1-基)-,(-)-(9CI)
乙基1,3-二氧戊环-4-羧酸酯
丙炔醛乙二醇缩醛
三甲基-[(2-甲基-1,3-二氧戊环-4-基)甲基]铵碘化物
三氟乙烯臭氧化物
三丁基(1,3-二恶烷-2-基甲基)溴化鏻
[2-(2-碘乙基)-1,3-二氧戊环-4-基]甲醇
6,8-二氧杂二螺[2.1.4.2]十一烷
6,7-二氧杂双环[3.2.1]辛-2-烯-8-羧酸
5H,8H-呋喃并[3,4:1,5]环戊二烯并[1,2-d]-1,3-二噁唑(9CI)
5-过氧化氢基-5-甲基-1,2-二恶烷-3-酮
5-嘧啶羧酸,4-(2-呋喃基)-1,2,3,4-四氢-6-甲基-2-羰基-,1-甲基乙基酯
5-(哌嗪-1-基)苯并呋喃-2-甲酰胺
5-(1,3-二氧杂烷-2-基)呋喃-2-磺酰氯
5-(1,3-二氧戊环-2-基)戊腈
5,5-二羟基戊醛
4a-乙基-2,4a,5,6,7,7a-六氢-4-(3-羟基苯基)-1-甲基-1H-1-吡喃并英并啶
4-硝基-4-丙基辛醛乙烯缩醛
4-甲基-4-硝基辛醛乙烯缩醛
4-甲基-2-戊基-1,3-二氧戊环
4-甲基-2-十一烷基-1,3-二氧戊环
4-甲基-2-[(1E)-1-戊烯-1-基]-1,3-二氧戊环
4-甲基-2-(三氯甲基)-1,3-二氧戊环
4-甲基-2-(2-(甲硫基)乙基)-1,3-二氧戊环
4-甲基-2-(1-丙烯基)-1,3-二氧戊环
4-甲基-1,3-二氧戊环
4-烯丙基-4-甲基-2-乙烯基-1,3-二氧戊环
4-溴-3,5,5-三甲基二氧戊环-3-醇
相关功能分类

联系我们



关注我们

公众号

