
8-苯基辛酸 | 26547-51-3

-
物化性质
-
计算性质
-
ADMET
-
安全信息
-
SDS
-
制备方法与用途
-
上下游信息
-
文献信息
-
表征谱图
-
同类化合物
-
相关功能分类
-
相关结构分类
物化性质
-
熔点:27 °C
-
沸点:150 °C
-
密度:1.020±0.06 g/cm3(Predicted)
-
闪点:150-152°C/0.1mm
-
稳定性/保质期:
- 如果遵照规格使用和储存,则不会分解。
- 避免接触氧化物。
- 存在于烟气中。
计算性质
-
辛醇/水分配系数(LogP):4.1
-
重原子数:16
-
可旋转键数:8
-
环数:1.0
-
sp3杂化的碳原子比例:0.5
-
拓扑面积:37.3
-
氢给体数:1
-
氢受体数:2
安全信息
-
危险品标志:Xi
-
危险类别码:R36/38
-
海关编码:2916399090
-
安全说明:S26,S36
-
危险性防范说明:P261,P264,P271,P280,P302+P352,P304+P340,P305+P351+P338,P312,P362,P403+P233,P501
-
危险性描述:H315,H319,H335
-
储存条件:保持贮藏器密封,并将其放入一个紧密的容器中。应储存在阴凉、干燥的地方。
SDS
上下游信息
-
上游原料
中文名称 英文名称 CAS号 化学式 分子量 6-苯基己酸甲酯 6-phenyl-hexanoic acid methyl ester 5581-76-0 C13H18O2 206.285 6-氧代-8-苯基庚酸 6-oxo-8-phenylheptanoic acid 53668-48-7 C14H18O3 234.295 苯已醇 6-phenyl-1-hexanol 2430-16-2 C12H18O 178.274 1-溴-6-苯基己烷 (6-bromohexyl)benzene 27976-27-8 C12H17Br 241.171 —— 8-phenyloct-5-enoic acid 85388-12-1 C14H18O2 218.296 -
下游产品
中文名称 英文名称 CAS号 化学式 分子量 —— methyl 8-phenyloctanoate 30884-34-5 C15H22O2 234.338 8-苯基-1-辛醇 8-phenyloctan-1-ol 10472-97-6 C14H22O 206.328 8-苯基辛酸乙烯酯 vinyl 8-phenyloctanoate 63339-71-9 C16H22O2 246.349 —— 8-phenyloctanamide 124400-98-2 C14H21NO 219.327 —— 8-phenyloctanoyl chloride 61875-56-7 C14H19ClO 238.757 7-苯甲酰基庚酸 8-oxo-8-phenyloctanoic acid 24314-23-6 C14H18O3 234.295 1-溴-8-苯基辛烷 1-bromo-8-phenyloctane 54646-75-2 C14H21Br 269.225 —— 7-Benzoyl-oenanthsaeure-methylester 94204-09-8 C15H20O3 248.322 —— 1,1,1-Trifluoro-9-phenyl-nonan-2-one —— C15H19F3O 272.31 —— N-(cyanomethyl)-8-phenyloctanamide 1010096-87-3 C16H22N2O 258.363
反应信息
-
作为反应物:参考文献:名称:3-(4-芳酰基-1-甲基-1H-2-吡咯基)-N-羟基-2-丙烯酰胺是一类新型的合成组蛋白脱乙酰基酶抑制剂。2.吡咯-C2和/或-C4取代对生物活性的影响。摘要:先前的SAR研究(第1部分:Mai,A .;等人,J。Med。Chem。2003,46,512-524)在3-(- 4-苯甲酰基-1-甲基-1H-吡咯-2-基)-N-羟基-2-丙烯酰胺(1a)突出显示了其4-苯基乙酰基(1b)和4-肉桂酰基(1c)类似物是抑制玉米的更有效化合物体外HD2活性。在本文中,我们研究了化学取代对1a-c的吡咯-C2乙烯链进行的化学取代对抗HD2活性的影响,这些链被亚甲基,乙烯,取代的乙烯和1,3-丁二烯链(化合物2)。生物学结果清楚地表明,未取代的乙烯链是获得最高HDAC抑制活性的最佳结构基序,唯一的例外是引入了1,3-丁二烯基部分变成1a化学结构(IC50(2f)= 0.77 microM; IC50(1a)= 3.8 microM)。化合物3的IC50值(制备为1b的同系物)显示,吡咯C-(4)位置的苯和羰基之间的APHA模板很好地接受了2至5个亚甲基的烃间DOI:10.1021/jm030990+
-
作为产物:描述:3-苯基-1-(噻吩-2-基)丙烷-1-酮 在 二硫化碳 、 sodium hydroxide 、 三氯化铝 、 aluminum nickel 、 alkaline aqueous sodium hypobromite solution 作用下, 生成 8-苯基辛酸参考文献:名称:Sy, Bulletin de la Societe Chimique de France, 1955, p. 1175,1178摘要:DOI:
文献信息
-
Novel Deoxyxylulosephosphate-Reductoisomerase Inhibitors: Fosmidomycin Derivatives with Spacious Acyl Residues作者:Regina Ortmann、Jochen Wiesner、Katrin Silber、Gerhard Klebe、Hassan Jomaa、Martin SchlitzerDOI:10.1002/ardp.200700149日期:2007.9enzyme of the mevalonate‐independent pathway of the isoprenoid biosynthesis. Using fosmidomycin as a specific inhibitor of Dxr, this enzyme was previously validated as target for the treatment of malaria and bacterial infections. The replacement of the formyl residue of fosmidomycin by spacious acyl residues yielded inhibitors active in the micromolar range. As predicted by flexible docking, evidence was
-
Inhibitors of squalene synthase and protein farnesyltransferase申请人:Abbott Laboratories公开号:US05831115A1公开(公告)日:1998-11-03The present invention provides a compound of the formula ##STR1## or a pharmaceutically acceptable salt thereof, which are useful for inhibiting protein farnesyltransferase and the farnesylation of the oncogene protein Ras or inhibiting de novo squalene production resulting in the inhibition of cholesterol biosynthesis, processes for the preparation of the compounds of the invention in addition to intermediates useful in these processes, a pharmaceutical composition, and to methods of using such compounds.
-
[EN] TOLL-LIKE RECEPTOR LIGANDS<br/>[FR] LIGANDS DU RÉCEPTEUR DE TYPE TOLL申请人:INIMMUNE CORP公开号:WO2019157509A1公开(公告)日:2019-08-15Toll-like receptor (TLR) ligands having an allose-based core are stable in aqueous formulation and are useful in treating, preventing, or reducing susceptibility to diseases or conditions mediated by TLRs, such as cancer, infectious disease, allergy, autoimmune disease, sepsis, and ischemia reperfusion.
-
Inhibition of monoamine oxidase by selected phenylalkylcaffeine analogues作者:Anél Petzer、Paul Grobler、Jacobus J Bergh、Jacobus P PetzerDOI:10.1111/jphp.12193日期:2014.4.11
Abstract Objectives Caffeine represents a useful scaffold for the design of monoamine oxidase (MAO) type B inhibitors. Specifically, substitution on the C8 position yields structures which are high-potency MAO-B inhibitors. To explore the structure–activity relationships of MAO-B inhibition by caffeine-derived compounds, this study examines the MAO inhibitory properties of a series of phenylalkylcaffeine analogues.
Methods Employing the recombinant human enzymes, the potencies (IC50 values) by which the caffeine analogues inhibit MAO-A and MAO-B were measured. The reversibility of inhibition of a selected inhibitor was determined by measuring the recovery of enzyme activity after dilution and dialysis of enzyme-inhibitor mixtures.
Key findings The results document that the phenylalkylcaffeine analogues are reversible and selective MAO-B inhibitors with a competitive mode of inhibition. The most potent analogue, 8-(7-phenylheptyl)caffeine, exhibits IC50 values for the inhibition of MAO-A and MAO-B of 3.01 μm and 0.086 μm, respectively. Increasing the length of the alkyl side chain leads to enhanced MAO-A and MAO-B inhibitory potency while introduction of a carbonyl group reduces MAO-B inhibitory potency.
Conclusions Phenylalkylcaffeines represent a new class of high-potency MAO-B inhibitors with the longer alkyl side chains yielding enhanced inhibitory activity. Such compounds may represent useful leads for the development of anti-parkinsonian therapies.
摘要 目标 咖啡因是设计单胺氧化酶(MAO)B型抑制剂的有用支架。特别是,C8位上的取代产生的高效MAO-B抑制剂的结构。为了探索咖啡因衍生物对MAO-B抑制的结构-活性关系,本研究检查了一系列苯基烷基咖啡因类似物的MAO抑制特性。
方法 使用重组人酶,测量咖啡因类似物抑制MAO-A和MAO-B的效力(IC50值)。通过测量酶-抑制剂混合物的稀释和透析后酶活性的恢复,确定了一种选定抑制剂的抑制可逆性。
关键发现 结果表明,苯基烷基咖啡因类似物是具有竞争性抑制模式的可逆和选择性MAO-B抑制剂。最有效的类似物,8-(7-苯基庚基)咖啡因,对MAO-A和MAO-B的抑制的IC50值分别为3.01 μm和0.086 μm。增加烷基侧链的长度可增强MAO-A和MAO-B的抑制效力,而引入羰基则降低MAO-B的抑制效力。
结论 苯基烷基咖啡因代表了一类新的高效MAO-B抑制剂,较长的烷基侧链可提高抑制活性。这类化合物可能成为开发抗帕金森疗法的有用先导。
-
Design, synthesis, and biological evaluation of oxazolidone derivatives as highly potent N-acylethanolamine acid amidase (NAAA) inhibitors作者:Jie Ren、Yuhang Li、Hongwei Ke、Yanting Li、Longhe Yang、Helin Yu、Rui Huang、Canzhong Lu、Yan QiuDOI:10.1039/c6ra28734d日期:——discovery of the oxazolidone derivative as a novel scaffold for NAAA inhibitors, and studied the structure–activity relationship (SAR) by modification of the side chain and terminal lipophilic substituents. The results showed that the link chain length of C5, straight and saturated linkages were the preferred shape patterns for NAAA inhibition. Several nanomolar NAAA inhibitors were described, includingN-乙酰乙醇胺水解酸酰胺酶(NAAA)是一种溶酶体酶,可催化内源性脂肪酸乙醇酰胺(FAE)(例如N-棕榈酰乙醇酰胺(PEA)。PEA通过与过氧化物酶体增殖物激活的受体α(PPAR-α)结合表现出抗炎和镇痛作用。已经提出通过抑制NAAA来防止PEA降解是治疗炎症和疼痛的新策略。在本研究中,我们报道了恶唑烷酮衍生物作为NAAA抑制剂的新型支架的发现,并通过修饰侧链和末端亲脂性取代基研究了结构-活性关系(SAR)。结果表明,C5的链长,直链和饱和键是抑制NAAA的优选形状。描述了几种纳摩尔NAAA抑制剂,包括2f,3h,3i和3jIC 50值分别为270 nM,150 nM,100 nM和190 nM。酶促降解研究表明2f以选择性,非竞争性和可逆的方式抑制NAAA。此外,在全身和口服给药后,2f表现出很高的抗炎和镇痛活性。
表征谱图
-
氢谱1HNMR
-
质谱MS
-
碳谱13CNMR
-
红外IR
-
拉曼Raman
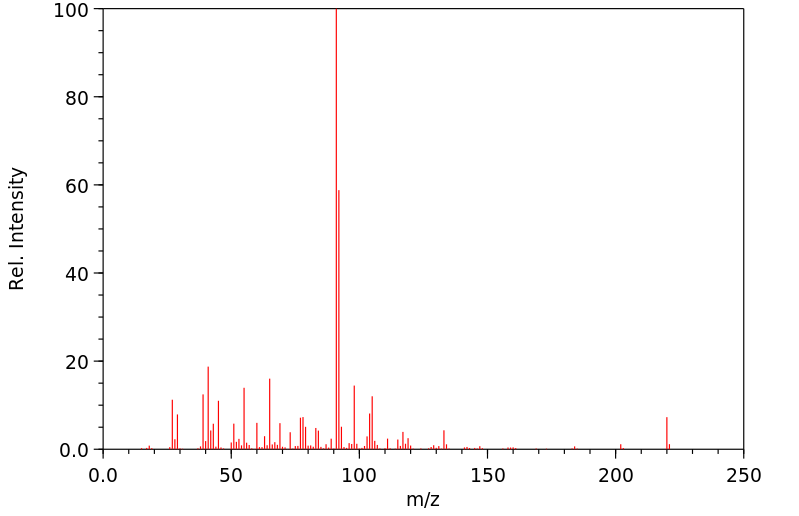
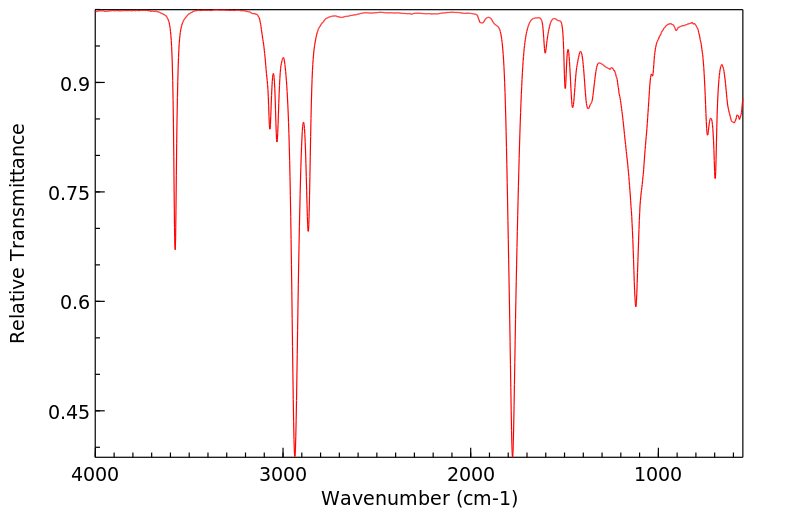
-
峰位数据
-
峰位匹配
-
表征信息
同类化合物





公众号
