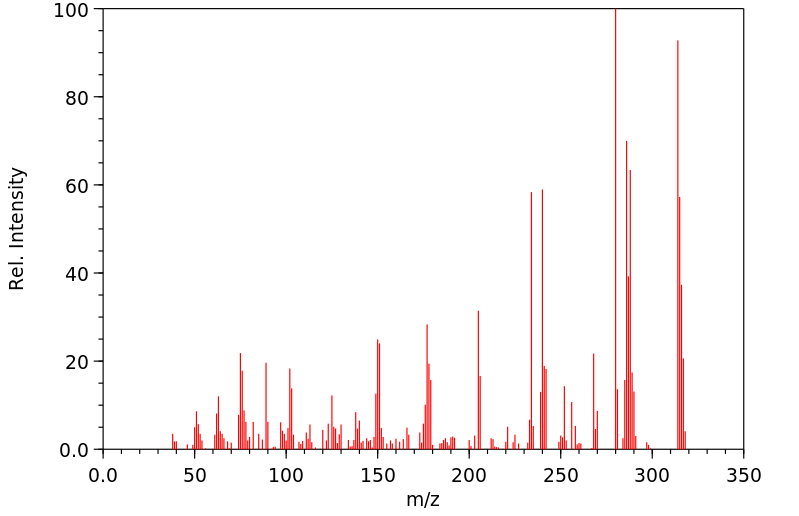
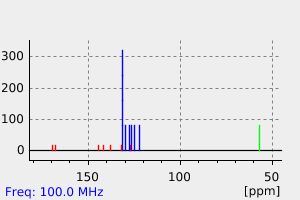
Clonazepam is metabolized principally in the liver. The metabolic pathways include hydroxylation, reduction of the nitro groups to amine groups, and the addition of acetate to the amino grouping. In particular, clonazepam is extensively metabolized by reduction to 7-amino-clonazepam and by N-acetylation to 7-acetamido-clonazepam. Hydroxylation at the C-3 position also occurs. Hepatic cytochrome P450 3A4 is implicated in the nitroreduction of clonazepam to pharmacologically inactive metabolites.
The shortcomings of clonazepam therapy include tolerance, withdrawal symptoms, and adverse effects such as drowsiness, dizziness, and confusion leading to increased risk of falls. Inter-individual variability in the incidence of adverse events in patients partly originates from the differences in clonazepam metabolism due to genetic and nongenetic factors. Since the prominent role in clonazepam nitro-reduction and acetylation of 7-amino-clonazepam is assigned to CYP3A and N-acetyl transferase 2 enzymes, respectively, the association between the patients' CYP3A status (CYP3A5 genotype, CYP3A4 expression) or N-acetyl transferase 2 acetylator phenotype and clonazepam metabolism (plasma concentrations of clonazepam and 7-amino-clonazepam) was evaluated in 98 psychiatric patients suffering from schizophrenia or bipolar disorders. The patients' CYP3A4 expression was found to be the major determinant of clonazepam plasma concentrations normalized by the dose and bodyweight (1263.5 +/- 482.9 and 558.5 +/- 202.4 ng/mL per mg/kg bodyweight in low and normal expressers, respectively, P<0.0001). Consequently, the dose requirement for the therapeutic concentration of clonazepam was substantially lower in low-CYP3A4 expresser patients than in normal expressers (0.029 +/- 0.011 vs 0.058 +/- 0.024 mg/kg bodyweight, P<0.0001). Furthermore, significantly higher (about 2-fold) plasma concentration ratio of 7-amino-clonazepam and clonazepam was observed in the patients displaying normal CYP3A4 expression and slower N-acetylation than all the others. Prospective assaying of CYP3A4 expression and N-acetyl transferase 2 acetylator phenotype can better identify the patients with higher risk of adverse reactions and can facilitate the improvement of personalized clonazepam therapy and withdrawal regimen.
Clonazepam is extensively metabolized in the liver to several metabolites including 7-aminoclonazepam, 7-acetaminoclonazepam, and 3-hydroxy derivatives of these metabolites and clonazepam. Clonazepam metabolites are excreted in urine by first-order kinetics, principally as their glucuronide and/or sulfate conjugates.
Hepatic (cytochrome P450, including CYP3A). Biotransformation occurs mainly by reduction of the 7-nitro group to the 4-amino derivative. This derivative can be acetylated, hydroxylated, and glucuronidated.
Route of Elimination: Clonazepam is highly metabolized, with less than 2% unchanged clonazepam being excreted in the urine. Metabolites of Klonopin are excreted by the kidneys. Clonazepam also undergoes acetylation via NAT2.
Half Life: 30-40 hours
IDENTIFICATION AND USE: Clonazepam is off-white to light yellow crystalline powder with a faint odor. It is a Schedule IV Controlled Substance. It is anticonvulsant and gamma-aminobutyric acid (GABA) modulator. A significant increase in the illegal use of clonazepam has been seen. HUMAN STUDIES: Overdosage of clonazepam may produce somnolence, confusion, ataxia, diminished reflexes, or coma. The shortcomings of clonazepam therapy include tolerance, withdrawal symptoms, and adverse effects such as drowsiness, dizziness, and confusion leading to increased risk of falls. Inter-individual variability in the incidence of adverse events in patients partly originates from the differences in clonazepam metabolism due to genetic and nongenetic factors. Because of experience with other members of the benzodiazepine class, clonazepam is assumed to be capable of causing an increased risk of congenital abnormalities when administered to a pregnant woman during the first trimester. In a study of 118 liveborn infants, there were 6 (5.1%) with major malformations, 4 of whom were prenatally exposed to clonazepam monotherapy and all of whom were full term infants. These included two with bilateral inguinal hernias requiring surgery, one with bilateral communicating hydroceles requiring surgery, one with midline pit over the nasal bridge, one with a uretero-pelvic junction obstruction requiring surgery and one with a PDA and ASD diagnosed at one day of age for which no follow-up is available. Sixty five (55.1%) of liveborn infants received a dysmorphological exam performed by an examiner who was blinded to the prenatal exposure. Although 16 (13.6%) children had 3 or more minor malformations, only one of the 16 was exposed to monotherapy. It is notable that 3 fullterm infants, all of whom were prenatally exposed to clonazepam monotherapy, had either inguinal hernias or communicating hydroceles requiring surgery. ANIMAL STUDIES: No adverse maternal or embryo-fetal effects were observed in mice and rats following oral administration during organogenesis of doses up to 15 mg/kg/day or 40 mg/kg/day, respectively. In a two-generation fertility study in which clonazepam was given orally to rats at 10 and 100 mg/kg/day, there was a decrease in the number of pregnancies and in the number of offspring surviving until weaning. In three studies in which clonazepam was administered orally to pregnant rabbits at doses of 0.2, 1, 5 or 10 mg/kg/day during the period of organogenesis, a similar pattern of malformations (cleft palate, open eyelid, fused sternebrae and limb defects) was observed in a low, non-dose-related incidence in exposed litters from all dosage groups. Reductions in maternal weight gain occurred at dosages of 5 mg/kg/day or greater and reduction in embryo-fetal growth occurred in one study at a dosage of 10 mg/kg/day. Of 1214 dogs exposed to clonazepam, 725 were symptomatic with 74% being ataxic, 33% sedated 21% agitated and 11% vomiting. Of 119 cats exposed to clonazepam, 85 were symptomatic with 85% being ataxic, 27% agitated and 27% sedated.
Allosteric interactions between central benzodiazepine receptors and gamma-aminobutyric acid (GABA) receptors potentiate the effects of GABA. As GABA is an inhibitory neurotransmitter, this results in increased inhibition of the ascending reticular activating system. Benzodiazepines, in this way, block the cortical and limbic arousal that occurs following stimulation of the reticular pathways.
Clonazepam, as with other benzodiazepines, is rarely associated with serum ALT elevations, and clinically apparent liver injury from clonazepam is extremely rare. However, at least one convincing case report of acute liver injury from clonazepam with recurrence on reexposure has been reported. Rare instances of drug induced liver injury has been reported with other benzodiazepines, such as chlordiazepoxide, diazepam, flurazepam, triazolam, clorazepate and alprazolam. In benzodiazepine related cases of acute liver injury, the latency has ranged from a few weeks to 6 months; the typical pattern of liver enzyme elevations has been cholestatic or mixed, but hepatocellular patterns have also been reported. The injury is usually mild to moderate in severity and self-limited. Fever and rash have not been described nor has autoantibody formation.
Clonazepam is rapidly and almost entirely absorbed after oral administration as tablets. Peak plasma concentrations of clonazepam administered by the oral route are reached within 1-4 hours and the associated absorption half-life is about 25 minutes. The absolute bioavailability is approximately 90% - but with substantially large differences between individuals.
Approximately 50-70% of a clonazepam dose is excreted in the urine and 10-30% is excreted in the feces as metabolites. The excretion of unchanged clonazepam in the urine is typically less than 2% of the administered dose. Metabolites of clonazepam are present in urine as both free and conjugated (glucuronide and sulfate) compounds.
Clonazepam distributes very rapidly to various organs and body tissues with preferential uptake by brain structures. The apparent volume of distribution has been documented as approximately 3 L/kg.
The documented clearance for clonazepam is approximately 55 ml/min regardless of gender. Nevertheless, clearance values normalized by weight decline with increasing body weight.
/MILK/ A 2750-g female infant was born at 36 weeks' gestation to a 40-year-old woman treated with clonazepam throughout her pregnancy. The infant developed apnea, cyanosis, and hypotonia within a few hours of birth. The mother's serum clonazepam level at delivery was 32 ng/mL; the cord blood level was 19 ng/mL. The infant had no congenital malformations, evidence of infection, or seizures. Clinical episodes ceased by ten days of age. The woman elected to breastfeed; breast milk clonazepam levels were between 11 and 13 ng/mL. She was discharged with a cardiorespiratory monitor. The authors suggest that infants of mothers receiving this agent during pregnancy or while nursing have serum levels measured. Additionally, these infants should be monitored for central nervous system depression or apnea.
[EN] S-NITROSOMERCAPTO COMPOUNDS AND RELATED DERIVATIVES<br/>[FR] COMPOSÉS DE S-NITROSOMERCAPTO ET DÉRIVÉS APPARENTÉS
申请人:GALLEON PHARMACEUTICALS INC
公开号:WO2009151744A1
公开(公告)日:2009-12-17
The present invention is directed to mercapto-based and S- nitrosomercapto-based SNO compounds and their derivatives, and their use in treating a lack of normal breathing control, including the treatment of apnea and hypoventilation associated with sleep, obesity, certain medicines and other medical conditions.
[EN] COMPOUNDS AND THEIR USE AS BACE INHIBITORS<br/>[FR] COMPOSÉS ET LEUR UTILISATION EN TANT QU'INHIBITEURS DE BACE
申请人:ASTRAZENECA AB
公开号:WO2016055858A1
公开(公告)日:2016-04-14
The present application relates to compounds of formula (I), (la), or (lb) and their pharmaceutical compositions/preparations. This application further relates to methods of treating or preventing Αβ-related pathologies such as Down's syndrome, β- amyloid angiopathy such as but not limited to cerebral amyloid angiopathy or hereditary cerebral hemorrhage, disorders associated with cognitive impairment such as but not limited to MCI ("mild cognitive impairment"), Alzheimer's disease, memory loss, attention deficit symptoms associated with Alzheimer's disease, neurodegeneration associated with diseases such as Alzheimer's disease or dementia, including dementia of mixed vascular and degenerative origin, pre-senile dementia, senile dementia and dementia associated with Parkinson's disease.
[EN] METHYL OXAZOLE OREXIN RECEPTOR ANTAGONISTS<br/>[FR] MÉTHYLOXAZOLES ANTAGONISTES DU RÉCEPTEUR DE L'OREXINE
申请人:MERCK SHARP & DOHME
公开号:WO2016089721A1
公开(公告)日:2016-06-09
The present invention is directed to methyl oxazole compounds which are antagonists of orexin receptors. The present invention is also directed to uses of the compounds described herein in the potential treatment or prevention of neurological and psychiatric disorders and diseases in which orexin receptors are involved. The present invention is also directed to compositions comprising these compounds. The present invention is also directed to uses of these compositions in the potential prevention or treatment of such diseases in which orexin receptors are involved.
Heterobicyclic compounds of Formula (I):
or a pharmaceutically-acceptable salt, tautomer, or stereoisomer thereof, as defined in the specification, and compositions containing them, and processes for preparing such compounds. Provided herein also are methods of treating disorders or diseases treatable by inhibition of PDE10, such as obesity, non-insulin dependent diabetes, schizophrenia, bipolar disorder, obsessive-compulsive disorder, Huntington's Disease, and the like.
Formula (I)的杂环化合物:
或其药用可接受的盐、互变异构体或立体异构体,如规范中所定义,并含有它们的组合物,以及制备这种化合物的方法。本文还提供了通过抑制PDE10来治疗由此可治疗的疾病或疾病的方法,如肥胖症、非胰岛素依赖型糖尿病、精神分裂症、躁郁症、强迫症、亨廷顿病等。
[EN] NAPHTHALENE CARBOXAMIDE M1 RECEPTOR POSITIVE ALLOSTERIC MODULATORS<br/>[FR] COMPOSÉS DE NAPHTHALÈNE CARBOXAMIDE, MODULATEURS ALLOSTÉRIQUES POSITIFS DU RÉCEPTEUR M1
申请人:MERCK SHARP & DOHME
公开号:WO2011149801A1
公开(公告)日:2011-12-01
The present invention is directed to naphthalene carboxamide compounds of formula (I) which are M1 receptor positive allosteric modulators and that are useful in the treatment of diseases in which the M1 receptor is involved, such as Alzheimers disease, schizophrenia, pain or sleep disorders. The invention is also directed to pharmaceutical compositions comprising the compounds and to the use of the compounds and compositions in the treatment of diseases mediated by the M1 receptor.