
吉拉尔特试剂 T | 123-46-6
中文名称
吉拉尔特试剂 T
中文别名
氯化三甲基铵乙酰肼;吉拉尔特试剂T;氯化甜菜碱酰肼;N-氯-三甲氨基乙酰肼;乙酰肼基三甲基氯化铵;乙酰肼三甲基氯化铵;吉腊德试剂T;(肼基羰基甲基)三甲胺氯化物;氯化三甲基铵乙酰胺;(羧甲基)三甲基氯化铵酰肼;2-肼基-N,N,N-三甲基-2-氧代乙铵氯化物
英文名称
girard's reagent T
英文别名
Girard’s reagent T;Girard T reagent;girard’s T reagent;(carboxymethyl)trimethylammonium chloride hydrazide;trimethylammoniumacetohydrazide chloride;2-hydrazinyl-N,N,N-trimethyl-2-oxoethan-1-aminium chloride;2-hydrazinyl-N,N,N-trimethyl-2-oxoethanaminium chloride;acethydrazide trimethylammonium chloride;Girard's T reagent;girard T;Girard reagent T;2-(Trimethylazaniumyl)ethanehydrazonate;hydrochloride;2-(trimethylazaniumyl)ethanehydrazonate;hydrochloride

CAS
123-46-6
化学式
C5H14N3O*Cl
mdl
MFCD00012009
分子量
167.639
InChiKey
YSULOORXQBDPCU-UHFFFAOYSA-N
BEILSTEIN
——
EINECS
——
-
物化性质
-
计算性质
-
ADMET
-
安全信息
-
SDS
-
制备方法与用途
-
上下游信息
-
文献信息
-
表征谱图
-
同类化合物
-
相关功能分类
-
相关结构分类
物化性质
-
熔点:188-192 °C (dec.) (lit.)
-
密度:1.3799 (rough estimate)
-
稳定性/保质期:
一、性质:白色针状结晶,具有较强的吸湿性。
计算性质
-
辛醇/水分配系数(LogP):-5.6
-
重原子数:10
-
可旋转键数:2
-
环数:0.0
-
sp3杂化的碳原子比例:0.8
-
拓扑面积:55.1
-
氢给体数:2
-
氢受体数:3
安全信息
-
安全说明:S16,S22,S24/25,S26,S36/37/39
-
危险品运输编号:NONH for all modes of transport
-
WGK Germany:3
-
海关编码:2928000090
-
危险品标志:Xn,F
-
危险类别码:R20/22,R44,R36/37/38,R11
-
RTECS号:CP3150000
-
危险性防范说明:P233,P260,P261,P264,P271,P280,P302+P352,P304,P304+P340,P305+P351+P338,P312,P321,P332+P313,P337+P313,P340,P362,P403,P403+P233,P405,P501
-
危险性描述:H315,H319,H335
-
储存条件:flammable area
SDS
模块 1. 化学品
1.1 产品标识符
: 吉拉尔特试剂 T
产品名称
1.2 鉴别的其他方法
Acethydrazide trimethylammonium chloride
(Carboxymethyl)trimethylammonium chloride hydrazide
(Hydrazinocarbonylmethyl)trimethylammonium chloride
1.3 有关的确定了的物质或混合物的用途和建议不适合的用途
仅用于研发。不作为药品、家庭或其它用途。
模块 2. 危险性概述
2.1 GHS-分类
根据全球协调系统(GHS)的规定,不是危险物质或混合物。
2.3 其它危害物 - 无
模块 3. 成分/组成信息
3.1 物 质
: Acethydrazide trimethylammonium chloride
别名
(Carboxymethyl)trimethylammonium chloride hydrazide
(Hydrazinocarbonylmethyl)trimethylammonium chloride
: C5H14ClN3O
分子式
: 167.64 g/mol
分子量
无
模块 4. 急救措施
4.1 必要的急救措施描述
吸入
如果吸入,请将患者移到新鲜空气处。 如呼吸停止,进行人工呼吸。
皮肤接触
用肥皂和大量的水冲洗。
眼睛接触
用水冲洗眼睛作为预防措施。
食入
切勿给失去知觉者通过口喂任何东西。 用水漱口。
4.2 主要症状和影响,急性和迟发效应
据我们所知,此化学,物理和毒性性质尚未经完整的研究。
4.3 及时的医疗处理和所需的特殊处理的说明和指示
无数据资料
模块 5. 消防措施
5.1 灭火介质
灭火方法及灭火剂
用水雾,抗乙醇泡沫,干粉或二氧化碳灭火。
5.2 源于此物质或混合物的特别的危害
碳氧化物, 氮氧化物, 氯化氢气体
5.3 给消防员的建议
如必要的话,戴自给式呼吸器去救火。
5.4 进一步信息
无数据资料
模块 6. 泄露应急处理
6.1 作业人员防护措施、防护装备和应急处置程序
避免粉尘生成。 避免吸入蒸气、烟雾或气体。
6.2 环境保护措施
不要让产品进入下水道。
6.3 泄漏化学品的收容、清除方法及所使用的处置材料
扫掉和铲掉。 放入合适的封闭的容器中待处理。
6.4 参考其他部分
丢弃处理请参阅第13节。
模块 7. 操作处置与储存
7.1 安全操作的注意事项
在有粉尘生成的地方,提供合适的排风设备。
7.2 安全储存的条件,包括任何不兼容性
贮存在阴凉处。 使容器保持密闭,储存在干燥通风处。
吸湿的.
7.3 特定用途
无数据资料
模块 8. 接触控制和个体防护
8.1 容许浓度
最高容许浓度
没有已知的国家规定的暴露极限。
8.2 暴露控制
适当的技术控制
常规的工业卫生操作。
个体防护设备
眼/面保护
请使用经官方标准如NIOSH (美国) 或 EN 166(欧盟) 检测与批准的设备防护眼部。
皮肤保护
所选择的保护手套必须符合EU的89/686/EEC规定和从它衍生出来的EN 376标准。
戴手套取 手套在使用前必须受检查。
请使用合适的方法脱除手套(不要接触手套外部表面),避免任何皮肤部位接触此产品.
使用后请将被污染过的手套根据相关法律法规和有效的实验室规章程序谨慎处理. 请清洗并吹干双手
身体保护
根据危险物质的类型,浓度和量,以及特定的工作场所选择身体保护措施。,
防护设备的类型必须根据特定工作场所中的危险物的浓度和数量来选择。
呼吸系统防护
不需要保护呼吸。如需防护粉尘损害,请使用N95型(US)或P1型(EN 143)防尘面具。
呼吸器使用经过测试并通过政府标准如NIOSH(US)或CEN(EU)的呼吸器和零件。
模块 9. 理化特性
9.1 基本的理化特性的信息
a) 外观与性状
形状: 结晶
颜色: 白色
b) 气味
无数据资料
c) 气味阈值
无数据资料
d) pH值
无数据资料
e) 熔点/凝固点
熔点/凝固点: 188 - 192 °C - 分解
f) 沸点、初沸点和沸程
无数据资料
g) 闪点
无数据资料
h) 蒸发速率
无数据资料
i) 易燃性(固体,气体)
无数据资料
j) 高的/低的燃烧性或爆炸性限度 无数据资料
k) 蒸气压
无数据资料
l) 蒸汽密度
无数据资料
m) 密度/相对密度
无数据资料
n) 水溶性
无数据资料
o) n-辛醇/水分配系数
无数据资料
p) 自燃温度
无数据资料
q) 分解温度
无数据资料
r) 粘度
无数据资料
模块 10. 稳定性和反应活性
10.1 反应性
无数据资料
10.2 稳定性
无数据资料
10.3 危险反应
无数据资料
10.4 应避免的条件
无数据资料
10.5 不相容的物质
强氧化剂
10.6 危险的分解产物
其它分解产物 - 无数据资料
模块 11. 毒理学资料
11.1 毒理学影响的信息
急性毒性
无数据资料
皮肤刺激或腐蚀
无数据资料
眼睛刺激或腐蚀
无数据资料
呼吸道或皮肤过敏
无数据资料
生殖细胞致突变性
无数据资料
致癌性
IARC:
此产品中没有大于或等于 0。1%含量的组分被 IARC鉴别为可能的或肯定的人类致癌物。
生殖毒性
无数据资料
特异性靶器官系统毒性(一次接触)
无数据资料
特异性靶器官系统毒性(反复接触)
无数据资料
吸入危险
无数据资料
潜在的健康影响
吸入 吸入可能有害。 可能引起呼吸道刺激。
摄入 如服入是有害的。
皮肤 通过皮肤吸收可能有害。 可能引起皮肤刺激。
眼睛 可能引起眼睛刺激。
接触后的征兆和症状
据我们所知,此化学,物理和毒性性质尚未经完整的研究。
附加说明
化学物质毒性作用登记: CP3150000
模块 12. 生态学资料
12.1 生态毒性
无数据资料
12.2 持久性和降解性
无数据资料
12.3 潜在的生物累积性
无数据资料
12.4 土壤中的迁移性
无数据资料
12.5 PBT 和 vPvB的结果评价
无数据资料
12.6 其它不良影响
无数据资料
模块 13. 废弃处置
13.1 废物处理方法
产品
将剩余的和不可回收的溶液交给有许可证的公司处理。
受污染的容器和包装
按未用产品处置。
模块 14. 运输信息
14.1 联合国危险货物编号
欧洲陆运危规: - 国际海运危规: - 国际空运危规: -
14.2 联合国运输名称
欧洲陆运危规: 非危险货物
国际海运危规: 非危险货物
国际空运危规: 非危险货物
14.3 运输危险类别
欧洲陆运危规: - 国际海运危规: - 国际空运危规: -
14.4 包裹组
欧洲陆运危规: - 国际海运危规: - 国际空运危规: -
14.5 环境危险
欧洲陆运危规: 否 国际海运危规 国际空运危规: 否
海洋污染物(是/否): 否
14.6 对使用者的特别提醒
无数据资料
参见发票或包装条的反面。
模块 15 - 法规信息
N/A
模块16 - 其他信息
N/A
制备方法与用途
反应信息
-
作为反应物:描述:参考文献:名称:水溶性有机锡 (IV) 与基于 Girard-T 试剂的腙的复合物:合成、光谱表征和抗菌活性摘要:两种离子腙,3-甲氧基水杨醛和 4-羟基水杨醛 Girard-T 肼氯化物,分别为 (3-OMeH2SalGT)Cl (1) 和 (4-OHH2SalGT)Cl (2),以及五种新的水溶性二有机锡配合物 R2Sn((3) -OMeSalGT)Cl [R = Ph (3), Me (4), Bu (5)] 和 R2Sn(4-OHSalGT) Cl [R = Ph (6), Me (7)] 已合成。光谱数据表明使用亚胺氮、烯醇和酚氧将配体去质子化并与锡配位为两性离子。配体和复合物的体外抗菌活性已针对革兰氏阳性菌(枯草芽孢杆菌和金黄色葡萄球菌)和革兰氏阳性菌进行了评估。阴性(大肠杆菌和铜绿假单胞菌)细菌。配体表现出低活性,而有机锡(IV)配合物表现出显着的抑制作用,并具有水溶性,这些化合物具有用作药物的极好潜力。DOI:10.1080/00958972.2013.809424
-
作为产物:参考文献:名称:羰基化合物的新形式,微量元素提取物和醛类缩醛的化学利用摘要:DOI:10.1002/hlca.193601901148
-
作为试剂:描述:(6R)-7t-(3,5-di-tert-butyl-4-hydroxy-benzylideneamino)-7c-methoxy-3-(1-methyl-1H-tetrazol-5-ylsulfanylmethyl)-8-oxo-(6rH)-5-thia-1-aza-bicyclo[4.2.0]oct-2-ene-2-carboxylic acid benzhydryl ester 在 吉拉尔特试剂 T 作用下, 以 甲醇 、 乙酸乙酯 为溶剂, 生成 7beta-氨基-7alpha-甲氧基-3-(1-甲基-1H-四唑-5-硫甲基)-8-氧代-5-硫-1-杂氮双环[4.2.0]辛-2-烯-2-甲酸二苯基甲酯参考文献:名称:A new semisynthetic 7.ALPHA.-methoxycephalosporin, CS-1170. 7.BETA.-(((cyanomethyl)thio)acetamido)-7.ALPHA.-methoxy-3-(((1-methyl-1H-tetrazol-5-yl)thio)methyl)-3-cephem-4-carboxylic acid.摘要:介绍了新半合成7α-甲氧基头孢菌素,7β-[[(氰甲基)硫代]乙酰胺基]-7α-甲氧基-3-[[(1-甲基-1H-四唑-5-基)硫代]甲基]-3-头孢烯-4-羧酸(CS-1170)的合成及其抗菌活性。与头孢西丁和头孢噻吩相比,该化合物显示出有趣的抗菌活性。DOI:10.7164/antibiotics.29.554
文献信息
-
13C-isotope labelling for the facilitated NMR analysis of a complex dynamic chemical system作者:Marta Dal Molin、Giulio Gasparini、Paolo Scrimin、Federico Rastrelli、Leonard J. PrinsDOI:10.1039/c1cc15295e日期:——13C-isotope labelling is presented as a novel tool for the study of complex chemical systems. 13C-isotope labelling permits the quantification of all 26 members of a dynamic library from a single 13C NMR spectrum without the need for advanced instrumentation or sophisticated experimental protocols.13C-同位素标记被提出作为一种研究复杂化学系统的新工具。13C-同位素标记允许通过单一的13C NMR谱图定量动态库中的所有26个成员,无需先进的仪器设备或复杂的实验方案。
-
Synthesis, Characterization, Catalytic Activity, and DFT Calculations of Zn(II) Hydrazone Complexes作者:Temiloluwa T. Adejumo、Nikolaos V. Tzouras、Leandros P. Zorba、Dušanka Radanović、Andrej Pevec、Sonja Grubišić、Dragana Mitić、Katarina K. Anđelković、Georgios C. Vougioukalakis、Božidar Čobeljić、Iztok TurelDOI:10.3390/molecules25184043日期:——theory (TD-DFT) at B3LYP/6-31G and B3LYP/6-311G(d,p) levels of theory. From the energies of frontier molecular orbitals (HOMO–LUMO), the reactivity descriptors, such as chemical potential (μ), hardness (η), softness (S), electronegativity (χ) and electrophilicity index (ω) have been calculated. The energetic behavior of the investigated compounds (1 and 2) has been examined in gas phase and solvent media合成了两种具有三齿腙基配体(2-乙酰基噻唑的缩合产物)的新型 Zn(II) 配合物,并通过红外 (IR) 和核磁共振 (NMR) 光谱以及单晶 X 射线衍射方法对其进行了表征。配合物 1、2 和最近合成的 [ZnL3(NCS)2] (L3 = (E)-N,N,N-trimethyl-2-oxo-2-(2-(1-(pyridin-2-yl)ethylidene) )肼基)乙-1-胺)配合物3作为酮-胺-炔(KA2)偶联反应的潜在催化剂进行了测试。已在密度泛函理论 (DFT)/B3LYP/6-31G 理论水平计算了新合成和表征的 Zn(II) 配合物的气相几何优化,而最高占据分子轨道和最低未占据分子轨道(HOMO 和 LUMO)能量是在 B3LYP/6-31G 和 B3LYP/6-311G(d,p) 水平的瞬态密度泛函理论 (TD-DFT) 内计算的的理论。从前沿分子轨道(HOMO-LUMO)
-
Spectroscopic studies of some thiosemicarbazide compounds derived from Girard's T and P作者:M.M. MostafaDOI:10.1016/j.saa.2006.02.063日期:2007.2in the isolated solid organic compounds have been determined using IR spectra. Also, the main functional groups in the skeleton of these compounds have been characterized using 1H NMR spectra. Moreover, a comparative studies between the three thiosemicarbazide derivatives have been carried out. The role of the existence of ethyl-, allyl-, phenyl-, [(CH3)3N+-] and [C5H5N+-] on the position of the functional
-
Synthesis of some novel CoII and CoIII complexes by tribochemical reactions using KI with some derivatives of thiosemicarbazide complexes derived from Girard's T and P作者:S.M. Al-Ashqar、M.M. MostafaDOI:10.1016/j.saa.2008.04.003日期:2008.12a tetrahedral geometry for all CoII complexes prepared by conventional chemical methods. The diamagnetic nature for three of the five iodide complexes, prepared by tribochemical reactions, suggests the oxidation of CoII to CoIII ion and the existence of low spin-octahedral geometry around the CoIII ion. Finally, the results of the rest of the iodide CoII complexes suggest either tetrahedral and/or high-spin通式为[Co(L1)2] Cl4.4 ,[Co(L1)CL2] Cl(L1 = N-([(烯丙基氨基)硫代甲基]肼基羰基甲基)三甲基氯化铵的起始Co(II)配合物),[Co(L2)Cl] Cl.2 。(1/2)EtOH(L2 = N-([[(乙胺)硫代甲基]肼基羰基甲基)三甲基氯化铵; ETHTC)和[Co(L3)CL2] Cl.2EtOH(通过常规化学方法合成L 3 = N-([((苯氨基甲基)硫代甲基]肼基羰基甲基)吡啶鎓氯化物; PTHPC)。通过用KI的化学方法获得的上述CoII配合物的摩擦化学反应提供了具有通式[Co(L1')I3。(1/2)EtOH] I,[Co2(L1')I4] I的新型CoII和CoIII配合物。 .EtOH,[Co(L2')I2。(3/2)EtOH] I,[Co2(L2')I4(OEt)2(H2O)2] I.(1/2)EtOH和[Co(L3' )I2
-
Synthesis, structures and magnetic properties of octahedral Co(III) complexes of heteroaromatic hydrazones with tetraisothiocyanato Co(II) anions作者:Božidar Čobeljić、Iztok Turel、Andrej Pevec、Zvonko Jagličić、Dušanka Radanović、Katarina Anđelković、Milica R. MilenkovićDOI:10.1016/j.poly.2018.08.070日期:2018.11complexes with heteroaromatic hydrazones ((E)-N,N,N-trimethyl-2-oxo-2-(2-(1-(pyridin-2-yl)ethylidene)hydrazinyl)ethan-1-aminium chloride and (E)-2-(1-(thiazol-2-yl)ethylidene)hydrazine-1-carbothioamide) and tetraisothiocyanato Co(II) complex anions have been synthesized, characterized by elemental analysis, IR and NMR spectroscopy, single-crystal X-ray diffraction and magnetic measurements. NMR spectroscopy
表征谱图
-
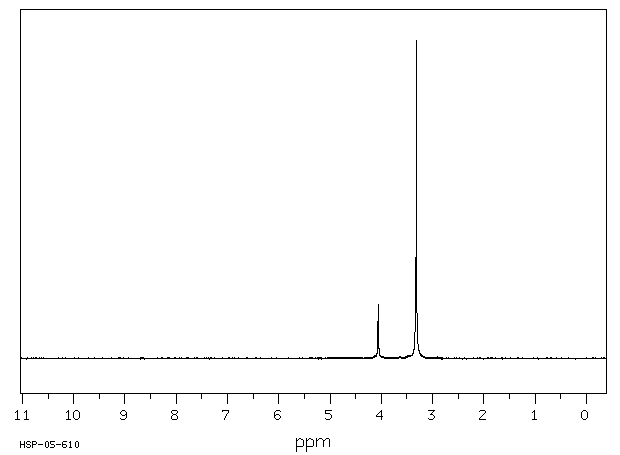
氢谱1HNMR
-
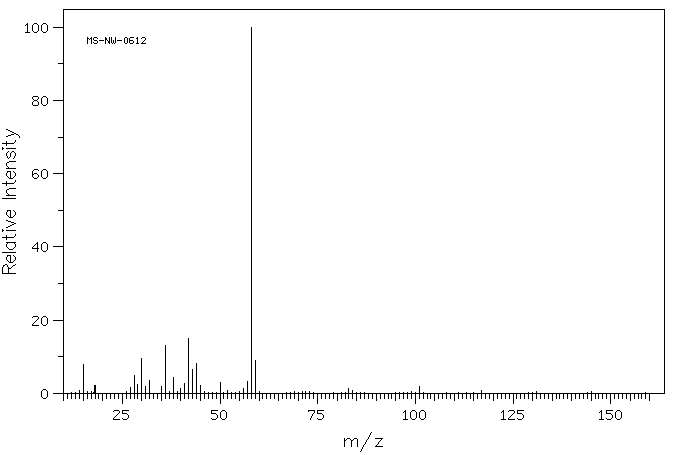
质谱MS
-
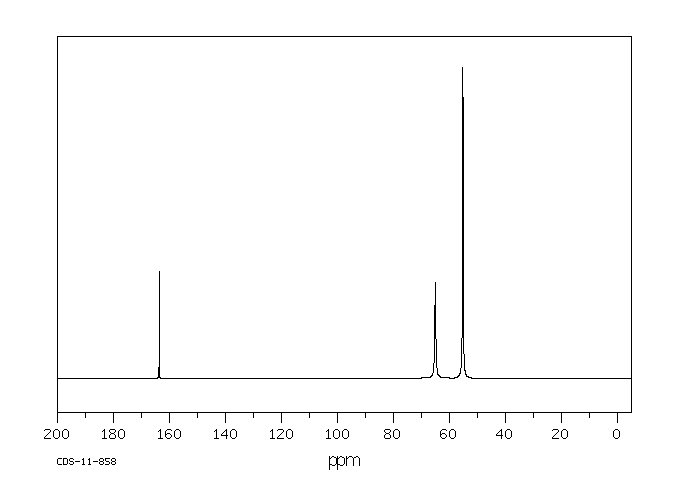
碳谱13CNMR
-
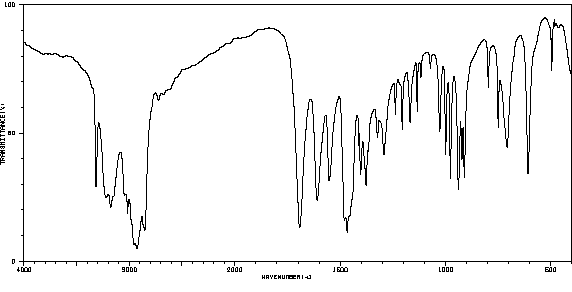
红外IR
-
拉曼Raman
-
峰位数据
-
峰位匹配
-
表征信息
同类化合物
(甲基3-(二甲基氨基)-2-苯基-2H-azirene-2-羧酸乙酯)
(±)-盐酸氯吡格雷
(±)-丙酰肉碱氯化物
(d(CH2)51,Tyr(Me)2,Arg8)-血管加压素
(S)-(+)-α-氨基-4-羧基-2-甲基苯乙酸
(S)-阿拉考特盐酸盐
(S)-赖诺普利-d5钠
(S)-2-氨基-5-氧代己酸,氢溴酸盐
(S)-2-[[[(1R,2R)-2-[[[3,5-双(叔丁基)-2-羟基苯基]亚甲基]氨基]环己基]硫脲基]-N-苄基-N,3,3-三甲基丁酰胺
(S)-2-[3-[(1R,2R)-2-(二丙基氨基)环己基]硫脲基]-N-异丙基-3,3-二甲基丁酰胺
(S)-1-(4-氨基氧基乙酰胺基苄基)乙二胺四乙酸
(S)-1-[N-[3-苯基-1-[(苯基甲氧基)羰基]丙基]-L-丙氨酰基]-L-脯氨酸
(R)-乙基N-甲酰基-N-(1-苯乙基)甘氨酸
(R)-丙酰肉碱-d3氯化物
(R)-4-N-Cbz-哌嗪-2-甲酸甲酯
(R)-3-氨基-2-苄基丙酸盐酸盐
(R)-1-(3-溴-2-甲基-1-氧丙基)-L-脯氨酸
(N-[(苄氧基)羰基]丙氨酰-N〜5〜-(diaminomethylidene)鸟氨酸)
(6-氯-2-吲哚基甲基)乙酰氨基丙二酸二乙酯
(4R)-N-亚硝基噻唑烷-4-羧酸
(3R)-1-噻-4-氮杂螺[4.4]壬烷-3-羧酸
(3-硝基-1H-1,2,4-三唑-1-基)乙酸乙酯
(2S,4R)-Boc-4-环己基-吡咯烷-2-羧酸
(2S,3S,5S)-2-氨基-3-羟基-1,6-二苯己烷-5-N-氨基甲酰基-L-缬氨酸
(2S,3S)-3-((S)-1-((1-(4-氟苯基)-1H-1,2,3-三唑-4-基)-甲基氨基)-1-氧-3-(噻唑-4-基)丙-2-基氨基甲酰基)-环氧乙烷-2-羧酸
(2S)-2,6-二氨基-N-[4-(5-氟-1,3-苯并噻唑-2-基)-2-甲基苯基]己酰胺二盐酸盐
(2S)-2-氨基-N,3,3-三甲基-N-(苯甲基)丁酰胺
(2S)-2-氨基-3-甲基-N-2-吡啶基丁酰胺
(2S)-2-氨基-3,3-二甲基-N-(苯基甲基)丁酰胺,
(2S)-2-氨基-3,3-二甲基-N-2-吡啶基丁酰胺
(2S,4R)-1-((S)-2-氨基-3,3-二甲基丁酰基)-4-羟基-N-(4-(4-甲基噻唑-5-基)苄基)吡咯烷-2-甲酰胺盐酸盐
(2R,3'S)苯那普利叔丁基酯d5
(2R)-2-氨基-3,3-二甲基-N-(苯甲基)丁酰胺
(2-氯丙烯基)草酰氯
(1S,3S,5S)-2-Boc-2-氮杂双环[3.1.0]己烷-3-羧酸
(1R,5R,6R)-5-(1-乙基丙氧基)-7-氧杂双环[4.1.0]庚-3-烯-3-羧酸乙基酯
(1R,4R,5S,6R)-4-氨基-2-氧杂双环[3.1.0]己烷-4,6-二羧酸
齐特巴坦
齐德巴坦钠盐
齐墩果-12-烯-28-酸,2,3-二羟基-,苯基甲基酯,(2a,3a)-
齐墩果-12-烯-28-酸,2,3-二羟基-,羧基甲基酯,(2a,3b)-(9CI)
黄酮-8-乙酸二甲氨基乙基酯
黄荧菌素
黄体生成激素释放激素(1-6)
黄体生成激素释放激素 (1-5) 酰肼
黄体瑞林
麦醇溶蛋白
麦角硫因
麦芽聚糖六乙酸酯
麦根酸
相关功能分类

联系我们



关注我们

公众号

