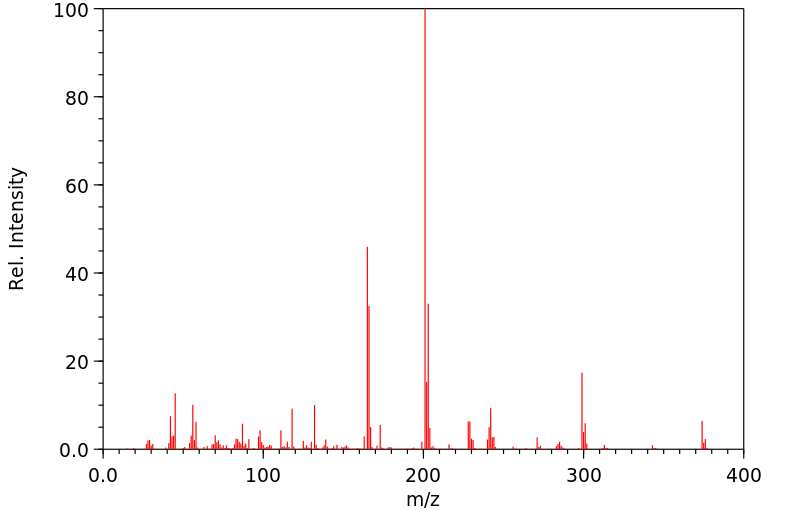
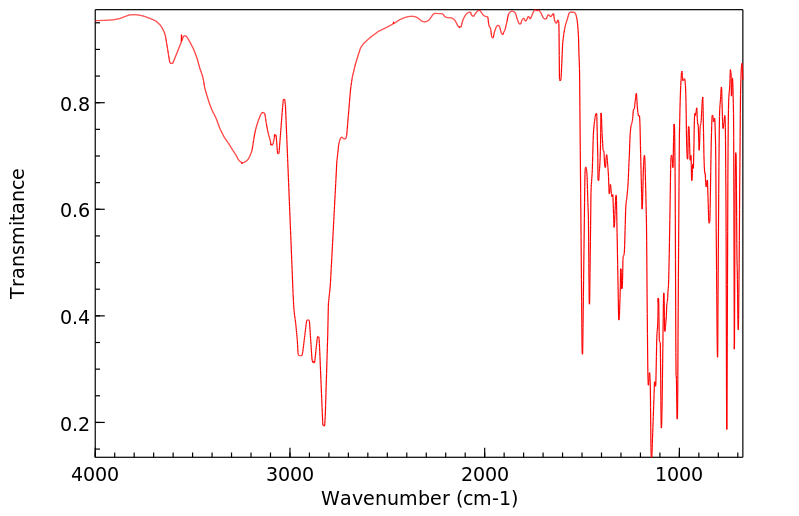
代谢
盐酸羟嗪在肝脏中通过CYP3A4和CYP3A5代谢。尽管
盐酸羟嗪的确切代谢命运尚不清楚,但其主要和活性代谢物(口服给药剂量的约45至60%),通过将其醇部分氧化为
羧酸生成,是第二代
抗组胺药[
西替利嗪]。
盐酸羟嗪可能分解为其他几种代谢物,尽管在人类中尚未阐明特定的结构和途径。
来源:DrugBank
代谢
氢化可的松及其活性代谢物
西替利嗪的药代动力学参数在给六只健康狗口服和静脉注射2毫克/千克
氢化可的松后确定。血浆药物
水平用高压
液相色谱法测定。药效学研究评估了对
组胺和抗犬IgE介导的皮肤风团形成的抑制效果。药代动力学和药效学相关性通过计算机建模确定。口服
氢化可的松的平均系统可用性为72%。无论给药途径如何,
氢化可的松都会迅速转化为
西替利嗪。在静脉和口服给药后,
西替利嗪的平均药时曲线下面积分别比
氢化可的松高8倍和10倍。口服给药
氢化可的松后,
西替利嗪的平均峰浓度约为2.2微克/毫升,而
氢化可的松为0.16微克/毫升。在静脉注射和口服给药
氢化可的松后,
西替利嗪的终末半衰期在10到11小时之间变化。将
西替利嗪血浆浓度与风团抑制数据进行比较,得到了S型关系。在最初的8小时内观察到最大抑制(
组胺和抗犬IgE介导的皮肤反应分别为82%和69%),这与
西替利嗪血浆浓度大于1.5微克/毫升相关。药理建模表明,增加
氢化可的松的剂量或给药频率不会导致比每日两次2毫克/千克的
氢化可的松更好的
组胺抑制作用。总之,
氢化可的松迅速转化为
西替利嗪。风团形成的减少几乎完全是由于
西替利嗪。药效学建模预测,每日两次口服2毫克/千克的
氢化可的松将产生最大的抗
组胺效果。
来源:Hazardous Substances Data Bank (HSDB)
代谢
尽管
羟嗪的确切代谢命运尚未明确建立,但看来该药物被完全代谢,主要在肝脏中进行。在动物中,
羟嗪及其代谢物通过胆汁消除在粪便中排出。
羟嗪的
羧酸代谢物,
西替利嗪,是一种长效
抗组胺药。
来源:Hazardous Substances Data Bank (HSDB)
代谢
肝脏
半衰期:20到25小时
来源:Toxin and Toxin Target Database (T3DB)
毒理性
盐酸羟嗪与
组胺竞争在效应细胞表面的H1受体位点,从而抑制
组胺引起的血管性
水肿、红斑和瘙痒。
盐酸羟嗪的镇静作用发生在中枢神经系统的皮层下
水平。由于其中枢抗
胆碱能作用,
盐酸羟嗪可能作为一种镇吐药有效。
来源:Toxin and Toxin Target Database (T3DB)
毒理性
尽管广泛使用,
羟嗪并未与肝功能测试异常或临床上明显的肝损伤有关联。实际上,
羟嗪常用于治疗与肝病相关的瘙痒。其安全性可能与每日低剂量和有限的使用期限有关。
可能性评分:E(不太可能是临床上明显肝损伤的原因)。
关于
抗组胺药的安全性和潜在肝毒性的参考资料,在
抗组胺药概述部分之后一起给出。
药物类别:
抗组胺药来源:LiverTox
毒理性
来源:Drug Induced Liver Injury Rank (DILIrank) Dataset
毒理性
DILI 注解:无 DILI(药物性肝损伤)担忧
来源:Drug Induced Liver Injury Rank (DILIrank) Dataset
毒理性
标签部分:没有匹配项
来源:Drug Induced Liver Injury Rank (DILIrank) Dataset
吸收、分配和排泄
氢化可的松的绝对
生物利用度尚未确定,因为静脉注射制剂由于溶血风险而不可用。
氢化可的松在口服给药后从胃肠道迅速吸收,大约在给药后2小时达到最大血浆浓度(Tmax)。
来源:DrugBank
吸收、分配和排泄
大约70%的
羟嗪活性代谢物,
西替利嗪,以不变的形式通过尿液排出。在人体中,肾和粪便排泄的确切程度尚未确定。
来源:DrugBank
吸收、分配和排泄
平均分布容积为16.0 ± 3.0 L/kg。在皮肤中的浓度高于血浆。
来源:DrugBank
吸收、分配和排泄
氢化可的松在儿童中的清除率为31.1 ± 11.1 mL/min/kg,在成人中为9.8 ± 3.3 mL/min/kg。
来源:DrugBank
吸收、分配和排泄
来源:Hazardous Substances Data Bank (HSDB)