
亚乙基丙二酸二甲酯 | 17041-60-0
中文名称
亚乙基丙二酸二甲酯
中文别名
——
英文名称
dimethyl ethylidenemalonate
英文别名
dimethyl 2-ethylidenemalonate;dimethyl 2-ethylidenepropanedioate

CAS
17041-60-0
化学式
C7H10O4
mdl
MFCD00008447
分子量
158.154
InChiKey
FRCFZWCJSXQAMQ-UHFFFAOYSA-N
BEILSTEIN
——
EINECS
——
-
物化性质
-
计算性质
-
ADMET
-
安全信息
-
SDS
-
制备方法与用途
-
上下游信息
-
文献信息
-
表征谱图
-
同类化合物
-
相关功能分类
-
相关结构分类
物化性质
-
沸点:154℃
-
密度:1.111 g/mL at 25 °C (lit.)
-
闪点:97 °C
-
稳定性/保质期:
按规定使用,这种物质不会分解,并且能避免氧化。
计算性质
-
辛醇/水分配系数(LogP):1
-
重原子数:11
-
可旋转键数:4
-
环数:0.0
-
sp3杂化的碳原子比例:0.428
-
拓扑面积:52.6
-
氢给体数:0
-
氢受体数:4
安全信息
-
海关编码:2917190090
-
WGK Germany:3
-
储存条件:存放在密闭、阴凉、干燥的地方即可。
SDS
1.1 产品标识符
: Dimethyl ethylidenemalonate
化学品俗名或商品名
1.2 鉴别的其他方法
无数据资料
1.3 有关的确定了的物质或混合物的用途和建议不适合的用途
仅供科研用途,不作为药物、家庭备用药或其它用途。
模块 2. 危险性概述
2.1 GHS分类
根据全球协调系统(GHS)的规定,不是危险物质或混合物。
当心 - 物质尚未完全测试。
2.3 其它危害物 - 无
模块 3. 成分/组成信息
3.1 物 质
: C7H10O4
分子式
: 158.15 g/mol
分子量
成分 浓度
Dimethyl ethylidenemalonate
-
17041-60-0
模块 4. 急救措施
4.1 必要的急救措施描述
如果吸入
如果吸入,请将患者移到新鲜空气处。 如果停止了呼吸,给于人工呼吸。
在皮肤接触的情况下
用肥皂和大量的水冲洗。
在眼睛接触的情况下
用水冲洗眼睛作为预防措施。
如果误服
切勿给失去知觉者从嘴里喂食任何东西。 用水漱口。
4.2 最重要的症状和影响,急性的和滞后的
4.3 及时的医疗处理和所需的特殊处理的说明和指示
无数据资料
模块 5. 消防措施
5.1 灭火介质
灭火方法及灭火剂
用水雾,耐醇泡沫,干粉或二氧化碳灭火。
5.2 源于此物质或混合物的特别的危害
碳氧化物
5.3 救火人员的预防
如必要的话,戴自给式呼吸器去救火。
5.4 进一步的信息
无数据资料
模块 6. 泄露应急处理
6.1 人员的预防,防护设备和紧急处理程序
防止吸入蒸汽、气雾或气体。
6.2 环境预防措施
不要让产物进入下水道。
6.3 抑制和清除溢出物的方法和材料
存放在合适的封闭的处理容器内。
6.4 参考其他部分
丢弃处理请参阅第13节。
模块 7. 操作处置与储存
7.1 安全操作的注意事项
一般性的防火保护措施。
7.2 安全储存的条件,包括任何不兼容性
贮存在阴凉处。 容器保持紧闭,储存在干燥通风处。
打开了的容器必须仔细重新封口并保持竖放位置以防止泄漏。
7.3 特定用途
无数据资料
模块 8. 接触控制/个体防护
8.1 控制参数
最高容许浓度
没有已知的国家规定的暴露极限。
8.2 暴露控制
适当的技术控制
常规的工业卫生操作。
人身保护设备
眼/面保护
请使用经官方标准如NIOSH (美国) 或 EN 166(欧盟) 检测与批准的设备防护眼部。
皮肤保护
戴手套取 手套在使用前必须受检查。
请使用合适的方法脱除手套(不要接触手套外部表面),避免任何皮肤部位接触此产品.
使用后请将被污染过的手套根据相关法律法规和有效的实验室规章程序谨慎处理. 请清洗并吹干双手
所选择的保护手套必须符合EU的89/686/EEC规定和从它衍生出来的EN 376标准。
身体保护
防渗透的衣服, 防护设备的类型必须根据特定工作场所中的危险物的浓度和含量来选择。
呼吸系统防护
不需要对呼吸系统保护.对少量挥发请采用美国OV/AG (US)标准类型的 或欧洲ABEK (EU EN
14387)标准类型的呼吸器过滤器.
呼吸器使用经过测试并通过政府标准如NIOSH(US)或CEN(EU)的呼吸器和零件。
模块 9. 理化特性
9.1 基本的理化特性的信息
a) 外观与性状
形状: 液体
b) 气味
无数据资料
c) 气味临界值
无数据资料
d) pH值
无数据资料
e) 熔点/凝固点
无数据资料
f) 起始沸点和沸程
无数据资料
g) 闪点
97.00 °C - 闭杯
h) 蒸发速率
无数据资料
i) 可燃性(固体,气体)
无数据资料
j) 高的/低的燃烧性或爆炸性限度 无数据资料
k) 蒸气压
无数据资料
l) 相对蒸气密度
无数据资料
m) 相对密度
1.111 g/mL 在 25 °C
n) 水溶性
无数据资料
o) 辛醇/水分配系数的对数值
无数据资料
p) 自燃温度
无数据资料
q) 分解温度
无数据资料
r) 粘度
无数据资料
模块 10. 稳定性和反应活性
10.1 反应性
无数据资料
10.2 化学稳定性
无数据资料
10.3 危险反应的可能性
无数据资料
10.4 避免接触的条件
无数据资料
10.5 不兼容的材料
强氧化剂
10.6 危险的分解产物
其它分解产物 - 无数据资料
模块 11. 毒理学资料
11.1 毒理学影响的信息
急性毒性
无数据资料
皮肤腐蚀/刺激
无数据资料
严重眼损伤 / 眼刺激
无数据资料
呼吸道或皮肤过敏
无数据资料
生殖细胞诱变
无数据资料
致癌性
IARC:
此产品中没有大于或等于 0。1%含量的组分被 IARC鉴别为可能的或肯定的人类致癌物。
生殖毒性
无数据资料
特异性靶器官系统毒性(一次接触)
无数据资料
特异性靶器官系统毒性(反复接触)
无数据资料
吸入危险
无数据资料
潜在的健康影响
吸入 吸入可能有害。 可能引起呼吸道刺激。
摄入 如服入是有害的。
皮肤 如果通过皮肤吸收可能是有害的。 可能引起皮肤刺激。
眼睛 可能引起眼睛刺激。
附加说明
化学物质毒性作用登记: 无数据资料
模块 12. 生态学资料
12.1 毒性
无数据资料
12.2 持久存留性和降解性
无数据资料
12.3 生物积累的潜在可能性
无数据资料
12.4 土壤中的迁移
无数据资料
12.5 PBT 和 vPvB的结果评价
无数据资料
12.6 其它不利的影响
无数据资料
模块 13. 废弃处置
13.1 废物处理方法
产品
污染了的包装物
作为未用过的产品弃置。
模块 14. 运输信息
欧洲陆运危规: - 国际海运危规: - 国际空运危规: -
14.2 联合国(UN)规定的名称
欧洲陆运危规: 无危险货物
国际海运危规: 无危险货物
国际空运危规: 无危险货物
14.3 运输危险类别
欧洲陆运危规: - 国际海运危规: - 国际空运危规: -
14.4 包裹组
欧洲陆运危规: - 国际海运危规: - 国际空运危规: -
14.5 环境危险
欧洲陆运危规: 否 国际海运危规 海运污染物: 否 国际空运危规: 否
14.6 对使用者的特别预防
无数据资料
模块 15 - 法规信息
N/A
模块16 - 其他信息
N/A
: Dimethyl ethylidenemalonate
化学品俗名或商品名
1.2 鉴别的其他方法
无数据资料
1.3 有关的确定了的物质或混合物的用途和建议不适合的用途
仅供科研用途,不作为药物、家庭备用药或其它用途。
模块 2. 危险性概述
2.1 GHS分类
根据全球协调系统(GHS)的规定,不是危险物质或混合物。
当心 - 物质尚未完全测试。
2.3 其它危害物 - 无
模块 3. 成分/组成信息
3.1 物 质
: C7H10O4
分子式
: 158.15 g/mol
分子量
成分 浓度
Dimethyl ethylidenemalonate
-
17041-60-0
模块 4. 急救措施
4.1 必要的急救措施描述
如果吸入
如果吸入,请将患者移到新鲜空气处。 如果停止了呼吸,给于人工呼吸。
在皮肤接触的情况下
用肥皂和大量的水冲洗。
在眼睛接触的情况下
用水冲洗眼睛作为预防措施。
如果误服
切勿给失去知觉者从嘴里喂食任何东西。 用水漱口。
4.2 最重要的症状和影响,急性的和滞后的
4.3 及时的医疗处理和所需的特殊处理的说明和指示
无数据资料
模块 5. 消防措施
5.1 灭火介质
灭火方法及灭火剂
用水雾,耐醇泡沫,干粉或二氧化碳灭火。
5.2 源于此物质或混合物的特别的危害
碳氧化物
5.3 救火人员的预防
如必要的话,戴自给式呼吸器去救火。
5.4 进一步的信息
无数据资料
模块 6. 泄露应急处理
6.1 人员的预防,防护设备和紧急处理程序
防止吸入蒸汽、气雾或气体。
6.2 环境预防措施
不要让产物进入下水道。
6.3 抑制和清除溢出物的方法和材料
存放在合适的封闭的处理容器内。
6.4 参考其他部分
丢弃处理请参阅第13节。
模块 7. 操作处置与储存
7.1 安全操作的注意事项
一般性的防火保护措施。
7.2 安全储存的条件,包括任何不兼容性
贮存在阴凉处。 容器保持紧闭,储存在干燥通风处。
打开了的容器必须仔细重新封口并保持竖放位置以防止泄漏。
7.3 特定用途
无数据资料
模块 8. 接触控制/个体防护
8.1 控制参数
最高容许浓度
没有已知的国家规定的暴露极限。
8.2 暴露控制
适当的技术控制
常规的工业卫生操作。
人身保护设备
眼/面保护
请使用经官方标准如NIOSH (美国) 或 EN 166(欧盟) 检测与批准的设备防护眼部。
皮肤保护
戴手套取 手套在使用前必须受检查。
请使用合适的方法脱除手套(不要接触手套外部表面),避免任何皮肤部位接触此产品.
使用后请将被污染过的手套根据相关法律法规和有效的实验室规章程序谨慎处理. 请清洗并吹干双手
所选择的保护手套必须符合EU的89/686/EEC规定和从它衍生出来的EN 376标准。
身体保护
防渗透的衣服, 防护设备的类型必须根据特定工作场所中的危险物的浓度和含量来选择。
呼吸系统防护
不需要对呼吸系统保护.对少量挥发请采用美国OV/AG (US)标准类型的 或欧洲ABEK (EU EN
14387)标准类型的呼吸器过滤器.
呼吸器使用经过测试并通过政府标准如NIOSH(US)或CEN(EU)的呼吸器和零件。
模块 9. 理化特性
9.1 基本的理化特性的信息
a) 外观与性状
形状: 液体
b) 气味
无数据资料
c) 气味临界值
无数据资料
d) pH值
无数据资料
e) 熔点/凝固点
无数据资料
f) 起始沸点和沸程
无数据资料
g) 闪点
97.00 °C - 闭杯
h) 蒸发速率
无数据资料
i) 可燃性(固体,气体)
无数据资料
j) 高的/低的燃烧性或爆炸性限度 无数据资料
k) 蒸气压
无数据资料
l) 相对蒸气密度
无数据资料
m) 相对密度
1.111 g/mL 在 25 °C
n) 水溶性
无数据资料
o) 辛醇/水分配系数的对数值
无数据资料
p) 自燃温度
无数据资料
q) 分解温度
无数据资料
r) 粘度
无数据资料
模块 10. 稳定性和反应活性
10.1 反应性
无数据资料
10.2 化学稳定性
无数据资料
10.3 危险反应的可能性
无数据资料
10.4 避免接触的条件
无数据资料
10.5 不兼容的材料
强氧化剂
10.6 危险的分解产物
其它分解产物 - 无数据资料
模块 11. 毒理学资料
11.1 毒理学影响的信息
急性毒性
无数据资料
皮肤腐蚀/刺激
无数据资料
严重眼损伤 / 眼刺激
无数据资料
呼吸道或皮肤过敏
无数据资料
生殖细胞诱变
无数据资料
致癌性
IARC:
此产品中没有大于或等于 0。1%含量的组分被 IARC鉴别为可能的或肯定的人类致癌物。
生殖毒性
无数据资料
特异性靶器官系统毒性(一次接触)
无数据资料
特异性靶器官系统毒性(反复接触)
无数据资料
吸入危险
无数据资料
潜在的健康影响
吸入 吸入可能有害。 可能引起呼吸道刺激。
摄入 如服入是有害的。
皮肤 如果通过皮肤吸收可能是有害的。 可能引起皮肤刺激。
眼睛 可能引起眼睛刺激。
附加说明
化学物质毒性作用登记: 无数据资料
模块 12. 生态学资料
12.1 毒性
无数据资料
12.2 持久存留性和降解性
无数据资料
12.3 生物积累的潜在可能性
无数据资料
12.4 土壤中的迁移
无数据资料
12.5 PBT 和 vPvB的结果评价
无数据资料
12.6 其它不利的影响
无数据资料
模块 13. 废弃处置
13.1 废物处理方法
产品
污染了的包装物
作为未用过的产品弃置。
模块 14. 运输信息
欧洲陆运危规: - 国际海运危规: - 国际空运危规: -
14.2 联合国(UN)规定的名称
欧洲陆运危规: 无危险货物
国际海运危规: 无危险货物
国际空运危规: 无危险货物
14.3 运输危险类别
欧洲陆运危规: - 国际海运危规: - 国际空运危规: -
14.4 包裹组
欧洲陆运危规: - 国际海运危规: - 国际空运危规: -
14.5 环境危险
欧洲陆运危规: 否 国际海运危规 海运污染物: 否 国际空运危规: 否
14.6 对使用者的特别预防
无数据资料
模块 15 - 法规信息
N/A
模块16 - 其他信息
N/A
上下游信息
-
下游产品
中文名称 英文名称 CAS号 化学式 分子量 —— dimethyl-2-(2-bromoethylidene)malonte 69231-27-2 C7H9BrO4 237.05
反应信息
-
作为反应物:描述:亚乙基丙二酸二甲酯 在 N-溴代丁二酰亚胺(NBS) 、 过氧化氢苯甲酰 作用下, 以 甲醇 、 四氯化碳 为溶剂, 反应 3.0h, 生成 1-cyano-2,2-diethoxycarbonyl cyclopropane参考文献:名称:Verhe, R.; De Kimpe, N.; De Buyck, L., Bulletin des Societes Chimiques Belges, 1983, vol. 92, # 4, p. 371 - 396摘要:DOI:
-
作为产物:参考文献:名称:Claus, Justus Liebigs Annalen der Chemie, 1878, vol. 191, p. 69,72,74摘要:DOI:
文献信息
-
A Unified Catalytic Asymmetric (4+1) and (5+1) Annulation Strategy to Access Chiral Spirooxindole‐Fused Oxacycles作者:Min Gao、Yanshu Luo、Qianlan Xu、Yukun Zhao、Xiangnan Gong、Yuanzhi Xia、Lin HuDOI:10.1002/anie.202105282日期:2021.9A unified catalytic asymmetric (N+1) (N=4, 5) annulation reaction of oxindoles with bifunctional peroxides has been achieved in the presence of a chiral phase-transfer catalyst (PTC). This general strategy utilizes peroxides as unique bielectrophilic four- or five-atom synthons to participate in the C−C and the subsequent umpolung C−O bond-forming reactions with one-carbon unit nucleophiles, thus providing
-
Stéréospécificité de l'addition de Michael d'ènethiolates avec les énones acycliques作者:Kafui Kpegba、Patrick Metzner、Rosé RakotonirinaDOI:10.1016/s0040-4020(01)80066-1日期:1989.1E-enones (3-penten-2-one, chalcone) occurs with high stereospecificity: anti/syn product ratios are identical to the cis/trans enethiolate ratios. A pseudo-cyclic transition state is proposed with substituents of the enone C-C double bond occupying favorable equatorial positions. The 1,4-addition reaction is kineticaly controlled. In contrast to ester enolates, enethiolates allow the use of enones without锂化二硫酯与无环 β 取代的 E 烯酮的 1,4-加成反应得到非对映异构体 5-氧代烷基硫代硫酸盐。通过化学相关性将反构型分配给主要的非对映异构体。这种立体化学取决于前手侧伴侣的构型。通过捕获实验分析中间烯硫酸盐的 cisl/trans 比率。观察到 2 例二硫酯的高选择性顺式去质子化。E-烯酮 (3-penten-2-one, chalcone) 的 Michael 添加具有很高的立体特异性:抗/syn 产物比率与顺式/反式烯乙酸盐比率相同。提出了一种伪循环过渡态,烯酮 C-C 双键的取代基占据有利的赤道位置。1,4-加成反应是动力学控制的。与酯烯醇酯相比,烯硫醇酯允许使用烯酮,而不需要叔丁基或苯基。观察到的非对映选择性(抗/合成比高达 95 : 5)提供了一种通过在非环系列中形成碳-碳键来创建两个邻近碳取代不对称中心的新方法。然而,当使用不饱和二酯作为受体时,观察到较差的选择性。
-
Reduction of Activated Alkenes by P <sup>III</sup> /P <sup>V</sup> Redox Cycling Catalysis作者:Lars Longwitz、Thomas WernerDOI:10.1002/anie.201912991日期:2020.2.10using a phosphetane oxide catalyst in the presence of a simple organosilane as the terminal reductant and water as the hydrogen source. Quantitative hydrogenation was observed when 1.0 mol % of a methyl‐substituted phosphetane oxide was employed as the catalyst. The procedure is highly selective towards activated double bonds, tolerating a variety of functional groups that are usually prone to reduction
-
Minimization of Back‐Electron Transfer Enables the Elusive sp <sup>3</sup> C−H Functionalization of Secondary Anilines作者:Huaibo Zhao、Daniele LeonoriDOI:10.1002/anie.202100051日期:2021.3.29Anilines are some of the most used class of substrates for application in photoinduced electron transfer. N,N‐Dialkyl‐derivatives enable radical generation α to the N‐atom by oxidation followed by deprotonation. This approach is however elusive to monosubstituted anilines owing to fast back‐electron transfer (BET). Here we demonstrate that BET can be minimised by using photoredox catalysis in the presence
-
Catalytic alkylation of remote C–H bonds enabled by proton-coupled electron transfer作者:Gilbert J. Choi、Qilei Zhu、David C. Miller、Carol J. Gu、Robert R. KnowlesDOI:10.1038/nature19811日期:2016.11unfunctionalized amides to direct the formation of new C–C bonds. Given the prevalence of amides in pharmaceuticals and natural products, we anticipate that this method will simplify the synthesis and structural elaboration of amine-containing targets. Moreover, this study demonstrates that concerted proton-coupled electron transfer can enable homolytic activation of common organic functional groups尽管在氢原子转移 (HAT) 催化方面取得了进展,但目前还没有能够均解 N-烷基酰胺的强氮氢 (N-H) 键的分子 HAT 催化剂。开发酰胺均裂方案的动机源于所得酰胺基自由基的效用,其参与各种合成有用的转化,包括烯烃胺化和定向碳氢 (C-H) 键功能化。在后一个过程中——经典的 Hofmann-Löffler-Freytag 反应的一个子集——酰胺自由基从未活化的脂肪族 C-H 键中去除氢原子。尽管功能强大,但这些转化通常需要对酰胺原料进行氧化 N-预官能化,以实现有效的酰胺基生成。而且,因为这些 N 活化基团经常被结合到最终产品中,这些方法通常不适合直接构建碳 - 碳(C-C)键。在这里,我们报告了一种通过质子耦合电子转移使 N-烷基酰胺的 N-H 键均裂来克服这些限制的方法。在该协议中,激发态铱光催化剂和弱磷酸盐碱协同作用,以协同的基本步骤从酰胺底物中去除质子和电子。所得的酰胺基自由基中间体被证明可以促进后续的
表征谱图
-
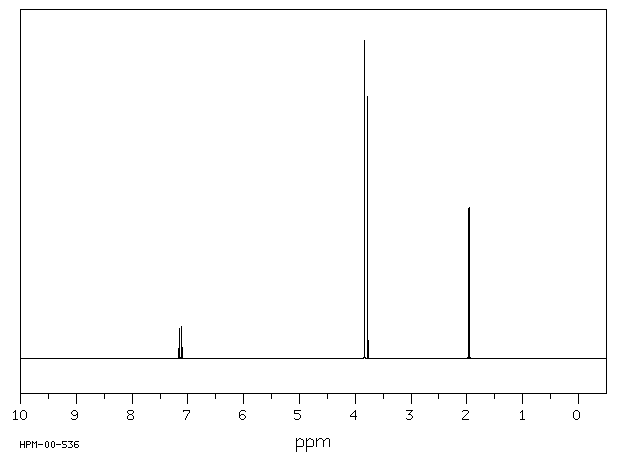
氢谱1HNMR
-
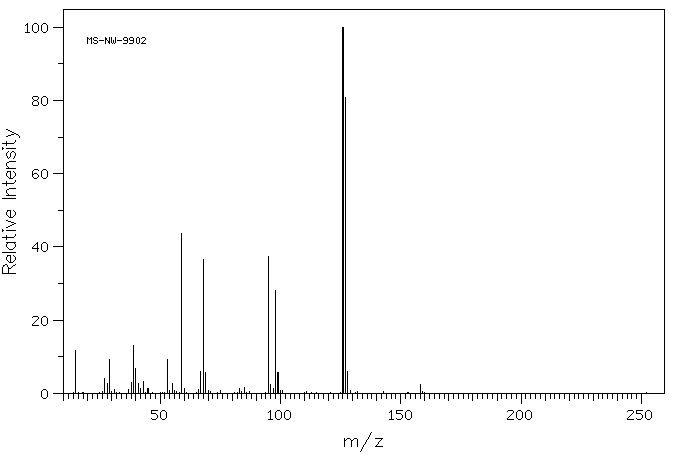
质谱MS
-
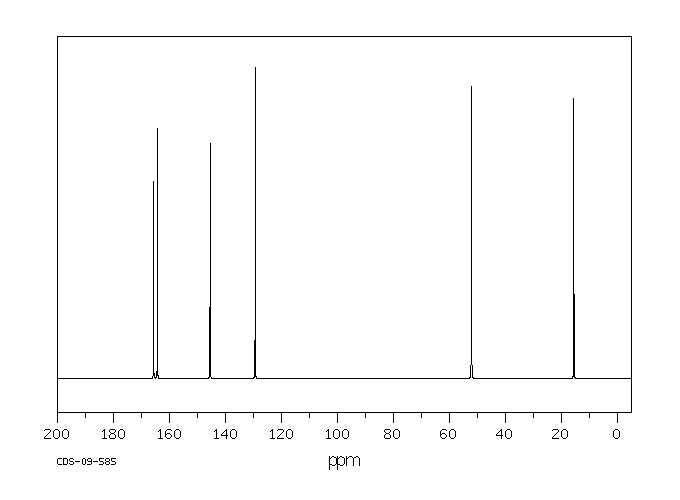
碳谱13CNMR
-
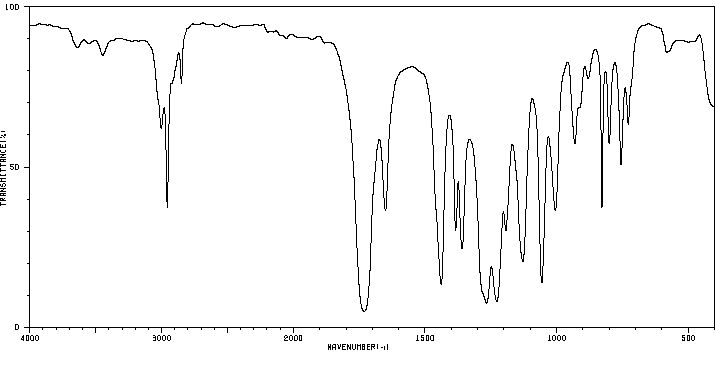
红外IR
-
拉曼Raman
-
峰位数据
-
峰位匹配
-
表征信息
同类化合物
(±)17,18-二HETE
(±)-辛酰肉碱氯化物
(Z)-5-辛烯甲酯
(Z)-4-辛烯酸
(R)-甲羟戊酸锂盐
(R)-普鲁前列素,游离酸
(R,R)-半乳糖苷
(E)-4-庚烯酸
(E)-4-壬烯酸
(E)-4-十一烯酸
(9Z,12E)-十八烷二烯酸甲酯
(6E)-8-甲基--6-壬烯酸甲基酯-d3
(3R,6S)-rel-8-[2-(3-呋喃基)-1,3-二氧戊环-2-基]-3-羟基-2,6-二甲基-4-辛酮
龙胆二糖
黑曲霉二糖
黄质霉素
麦芽酮糖一水合物
麦芽糖醇
麦芽糖酸
麦芽糖基蔗糖
麦芽糖一水合物
麦芽糖
鳄梨油酸乙酯
鲸蜡醇蓖麻油酸酯
鲸蜡醇油酸酯
鲸蜡硬脂醇硬脂酸酯
鲸蜡烯酸脂
鲸蜡基花生醇
鲫鱼酸
鲁比前列素
鲁比前列素
高级烷基C16-18-醇
高甲羟戊酸
高效氯氰菊酯
高-gamma-亚油酸
马来酸烯丙酯
马来酸氢异丙酯
马来酸氢异丁酯
马来酸氢丙酯
马来酸氢1-[2-(2-羟基乙氧基)乙基]酯
马来酸单乙酯
马来酸单丁酯
马来酸二辛酯
马来酸二癸酯
马来酸二甲酯
马来酸二烯丙酯
马来酸二正丙酯
马来酸二戊基酯
马来酸二异壬酯
马来酸二异丙酯

联系我们



关注我们

公众号

