
氯二氟乙酰胺 | 354-28-9

-
物化性质
-
计算性质
-
ADMET
-
安全信息
-
SDS
-
制备方法与用途
-
上下游信息
-
文献信息
-
表征谱图
-
同类化合物
-
相关功能分类
-
相关结构分类
物化性质
-
熔点:79-81 °C(lit.)
-
沸点:93°C 18mm
-
密度:1.529±0.06 g/cm3(Predicted)
计算性质
-
辛醇/水分配系数(LogP):0.5
-
重原子数:7
-
可旋转键数:1
-
环数:0.0
-
sp3杂化的碳原子比例:0.5
-
拓扑面积:43.1
-
氢给体数:1
-
氢受体数:3
安全信息
-
TSCA:Yes
-
危险品标志:Xn,Xi
-
危险类别码:R36/37/38
-
WGK Germany:3
-
海关编码:2924199090
-
安全说明:S26,S36
-
储存条件:室温
SDS
反应信息
-
作为反应物:参考文献:名称:2-氯-2,2-二氟乙酰胺(ClF2CC(O)NH2)。热分解,蒸气红外,质谱,低温NMR和理论研究。溶剂对构象偏好的影响摘要:在270至290°C的温度下研究了2-氯-2,2-二氟乙酰胺(CDFA)的气相热分解。分解的速率常数遵循Arrhenius方程。 质谱分析标题化合物的分解图。记录了质子和非质子溶剂中气相的FT-IR光谱和CDFA的红外光谱。通过使用6-31G *,6-31 + G *,6-311 + G **,aug-cc-pVDZ和6-31G *在密度泛函理论水平上进行的理论计算(PBEPBE和B3LYP方法)研究了势能面。 aug-cc-pVTZ基集。DOI:10.1071/ch10263
-
作为产物:描述:参考文献:名称:Reactions of fluoro olefins摘要:DOI:10.1007/bf00942892
-
作为试剂:描述:7a-(1,4-dicarbonylpentenyl)-9(11)-encanrenone 在 dipotassium hydrogenphosphate 、 氯二氟乙酰胺 、 双氧水 、 potassium hydrogencarbonate 、 臭氧 作用下, 以 四氢呋喃 、 二氯甲烷 、 异丙醇 、 甲苯 为溶剂, 生成 依普利酮参考文献:名称:一种高效低污染的依普利酮制备方法摘要:本发明公开了一种高效低污染的依普利酮制备方法,属于甾体激素药物的制备加工技术领域。该方法以化合物Ⅰ即Δ9,11‑坎利酮为起始原料,经7位呋喃基加成、呋喃基开环、内酯化成环反应得到5,7‑内酯,再经开环甲基化、9,11位双键环氧化反应制得依普利酮。本发明方法的原料和试剂价廉易得,在设备投入和生产成本上有极高的市场竞争力;所用试剂环境污染小,特别是不使用含氰基类高毒化合物,更加绿色环保;生产工艺操作易控制,收率高,成本低,适合工业化生产。公开号:CN111848719A
文献信息
-
Haloguanidine compounds, pharmaceutical compositions and methods of use申请人:ICI Americas Inc.公开号:US04362728A1公开(公告)日:1982-12-07Compounds useful for inhibiting gastric acid secretion and for the treatment of peptic ulcers caused or exacerbated by gastric acidity having the following formula (I): ##STR1## in which R.sup.1 and R.sup.2, are H, C.sub.1-10 alkyl, C.sub.3-8 cycloalkyl or cycloalkylalkyl in which the alkyl part is C.sub.1-6 and the cycloalkyl part is C.sub.3-8, each of the alkyl, cycloalkyl and cycloalkylalkyls being optionally substituted by one or more halogens selected from F, Cl and Br, provided that at least one of R.sup.1 and R.sup.2 is a halogen substituted alkyl, cycloalkyl or cycloalkylalkyl and provided that there is no halogen substituent on the carbon directly attached to the nitrogen; and X, m, Y, n and R.sup.3 are as described in the specification; and the pharmaceutically-acceptable acid-addition salts thereof. Processes for producing compounds of formula (I), pharmaceutical compositions containing them, methods of utilizing such compositions and intermediates useful for synthesizing compounds of formula (I) are also described.用于抑制胃酸分泌和治疗由胃酸引起或加重的消化性溃疡的化合物具有以下结构式(I):##STR1##其中R.sup.1和R.sup.2为H,C.sub.1-10烷基,C.sub.3-8环烷基或环烷基烷基,其中烷基部分为C.sub.1-6,环烷基部分为C.sub.3-8,每个烷基,环烷基和环烷基烷基可选择地被来自F、Cl和Br的一个或多个卤素取代,前提是R.sup.1和R.sup.2中至少有一个是卤素取代的烷基,环烷基或环烷基烷基,并且直接连接到氮的碳上没有卤素取代物;X,m,Y,n和R.sup.3如规范中所述;以及其药用可接受的酸加盐。还描述了制备结构式(I)化合物的方法,含有它们的药物组合物,利用这种组合物的方法以及用于合成结构式(I)化合物的中间体。
-
Allyl, epoxy and glycosyl perfluoroimidates. One-pot preparation and reaction作者:Noriyuki Nakajima、Miho Saito、Masabumi Kudo、Makoto UbukataDOI:10.1016/s0040-4020(02)00305-8日期:2002.4obtained trifluoroacetimidates were more stable than the trichloro analogue and were easily purified by SiO2 column chromatography and/or distillation. The 3,3-sigmatropic rearrangement of allylic analogues, acid-catalyzed cyclization of the epoxy analogues and glycosylation of sugar analogues were studied comparing with their corresponding trichloroacetimidates. The trifluoroacetimidates were considerably
-
Synthesis and structure-activity relationships of benzophenone hydrazone derivatives with insecticidal activity作者:Manfred B�ger、Dieter D�rr、Laurenz Gsell、Roger G Hall、Friedrich Karrer、Odd Kristiansen、Peter Maienfisch、Alfons Pascual、Alfred RindlisbacherDOI:10.1002/1526-4998(200102)57:2<191::aid-ps275>3.0.co;2-o日期:2001.2A broad range of benzophenone hydrazone derivatives was prepared and tested against selected chewing insect pests, allowing the analysis of structure-activity relationships. Good activity was found only when the aromatic rings were substituted at the 4-positions with an halogen atom and a triflate or perhaloalkoxy group. In contrast, a number of substituents on the hydrazone part led to active compounds
-
Novel Heteroaromatic Amides And Thioamides As Pesticides申请人:Bretschneider Thomas公开号:US20110098287A1公开(公告)日:2011-04-28The present application relates to novel amides and thioamides, to processes for preparation thereof and to use thereof for controlling animal pests, in particular arthropods and especially insects.本申请涉及新型酰胺和硫代酰胺,以及其制备过程和用于控制动物害虫,特别是节肢动物,尤其是昆虫的用途。
-
Three new [<i>X</i>C(O)NH]P(O)[N(CH<sub>2</sub>C<sub>6</sub>H<sub>5</sub>)<sub>2</sub>]<sub>2</sub>phosphoric triamides (<i>X</i>= CClF<sub>2</sub>, 3-F-C<sub>6</sub>H<sub>4</sub>and 3,5-F<sub>2</sub>-C<sub>6</sub>H<sub>3</sub>): a database analysis of tertiary N-atom geometry in compounds with a C(O)NHP(O)[N]<sub>2</sub>core作者:Mehrdad Pourayoubi、Jerry P. Jasinski、Samad Shoghpour Bayraq、Hossein Eshghi、Amanda C. Keeley、Giuseppe Bruno、Hadi Amiri RudbariDOI:10.1107/s0108270112036396日期:2012.10.15
In the phosphoric triamides
N ,N ,N ′,N ′-tetrabenzyl-N ′′-(2-chloro-2,2-difluoroacetyl)phosphoric triamide, C30H29ClF2N3O2P, (I),N ,N ,N ′,N ′-tetrabenzyl-N ′′-(3-fluorobenzoyl)phosphoric triamide, C35H33FN3O2P, (II), andN ,N ,N ′,N ′-tetrabenzyl-N ′′-(3,5-difluorobenzoyl)phosphoric triamide, C35H32F2N3O2P, (III), the tertiary N atoms of the dibenzylamido groups havesp 2character with minimal deviation from planarity. The sums of the three bond angles about the N atoms in (I)–(III) deviate by less than 8° from the planar value of 360°. The geometries of the tertiary N atoms in all phosphoric triamides with C(O)NHP(O)[N]2skeletons deposited in the Cambridge Structural Database [CSD; Allen (2002).Acta Cryst. B58 , 380–388] have been examined and the bond-angle sums at the two tertiary N atoms (SUM1 and SUM2) and the parameter ΔSUM (= SUM1 − SUM2) considered. It was found that in compounds with a considerable ΔSUM value, the more pyramidal N atoms are usually oriented so that the corresponding lone electron pair isanti with respect to the P=O group. In (I), (II) and (III), the phosphoryl and carbonyl groups, separated by an N atom, areanti with respect to each other. In the C(O)NHP(O) fragment of (I)–(III), the P—N bond is longer and the O—P—N angle is contracted compared with the other two P—N bonds and the O—P—N angles in the molecules. These effects are also seen in analogous compounds deposited in the CSD. Compounds with [C(O)NH]P(O)[N]X (X ≠ N), such as compounds with a [C(O)NH]P(O)[N][O] skeleton, have not been considered here. Also, compounds with a [C(O)NH]2P(O)[N] fragment have not been reported to date. In the crystal structures of all three title compounds, adjacent molecules are linkedvia pairs of P=O...H—N hydrogen bonds, forming dimers withC i symmetry.在磷酸三酰胺N,N,N′,N′-四苄基-N′′-(2-氯-2,2-二氟乙酰基)磷酸三酰胺中、(I)、N,N,N′,N′-四苄基-N′′-(3-氟苯甲酰基)磷酸三酰胺,C35H33FN3O2P、(II)和 N,N,N′,N′-四苄基-N′′-(3,5-二氟苯甲酰基)磷酸三酰胺 C35H32F2N3O2P,(III),二苄基氨基的三级 N 原子的平面偏离最小。(I)-(III)中关于 N 原子的三个键角之和与 360°的平面值偏差小于 8°。对剑桥结构数据库[CSD; Allen (2002).Acta Cryst.B58, 380-388]中所有具有 C(O)NHP(O)[N]2 骨架的磷酸三酰胺中三级 N 原子的几何形状进行了研究,并考虑了两个三级 N 原子(SUM1 和 SUM2)上的键角总和以及参数 ΔSUM (= SUM1 - SUM2)。研究发现,在具有较大 ΔSUM 值的化合物中,较多金字塔形 N 原子的取向通常使相应的孤电子对与 P=O 基团成反方向。在(I)、(II)和(III)中,被一个 N 原子隔开的磷酰基和羰基相互反向。在(I)-(III)的 C(O)NHP(O)片段中,与分子中的其他两个 P-N 键和 O-P-N 角相比,P-N 键较长,O-P-N 角收缩。这些效应在沉积在 CSD 中的类似化合物中也可以看到。这里没有考虑[C(O)NH]P(O)[N]X(X≠N)的化合物,例如具有[C(O)NH]P(O)[N][O]骨架的化合物。此外,含有 [C(O)NH]2P(O)[N] 片段的化合物至今也未见报道。在所有这三种标题化合物的晶体结构中,相邻分子都以 P=O...H-N 氢键成对连接,形成具有对称性的二聚体。
表征谱图
-
氢谱1HNMR
-
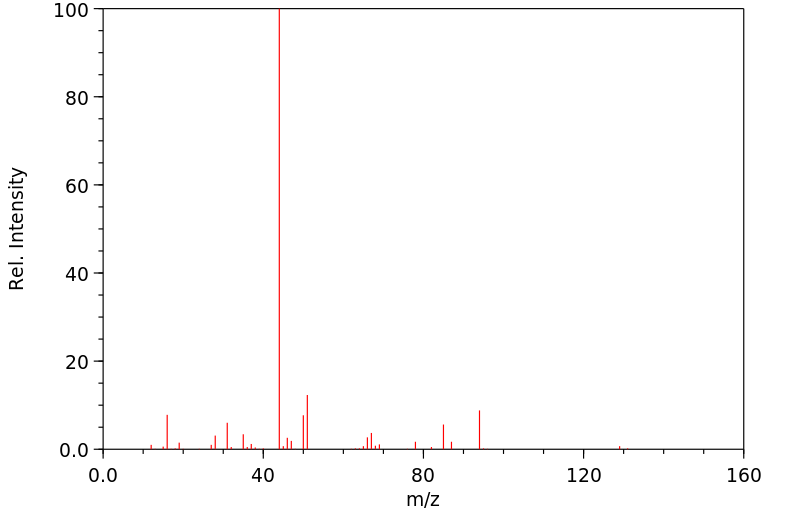
质谱MS
-
碳谱13CNMR
-
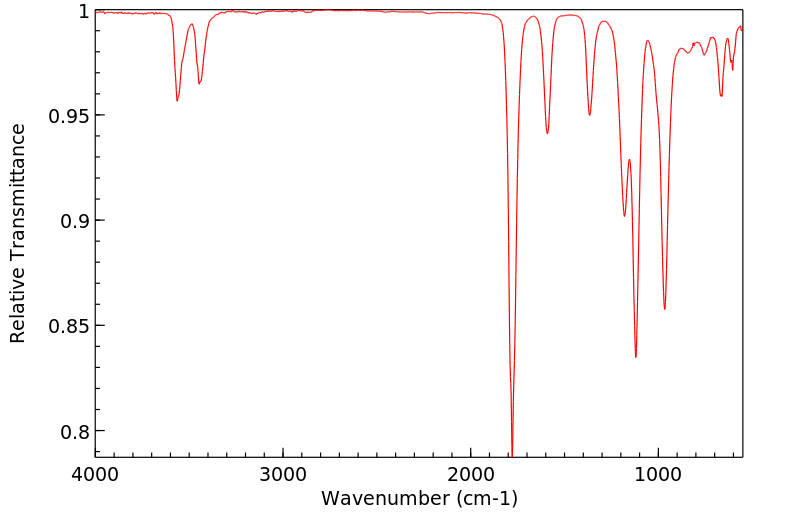
红外IR
-
拉曼Raman
-
峰位数据
-
峰位匹配
-
表征信息
同类化合物





公众号

