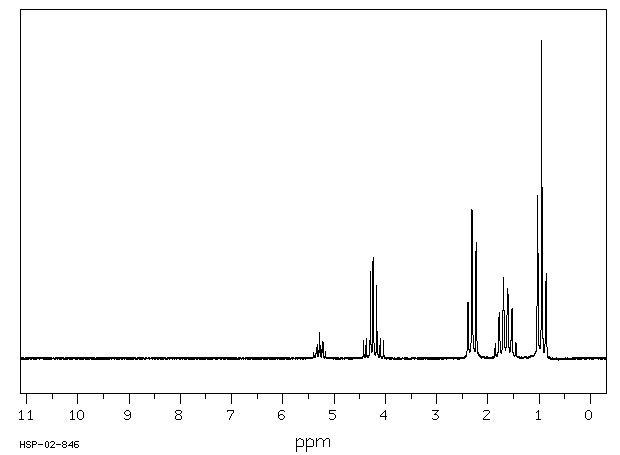
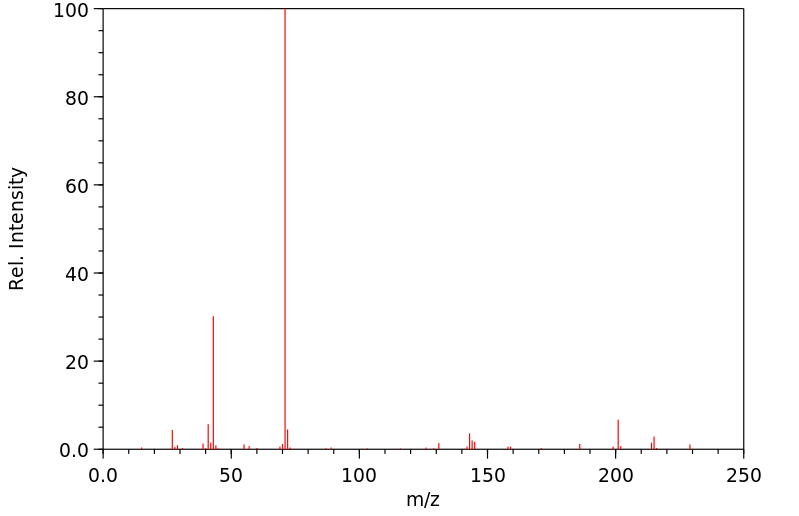
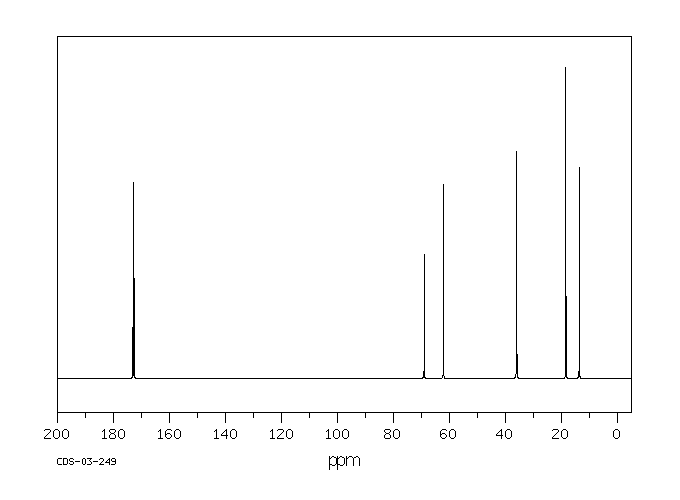
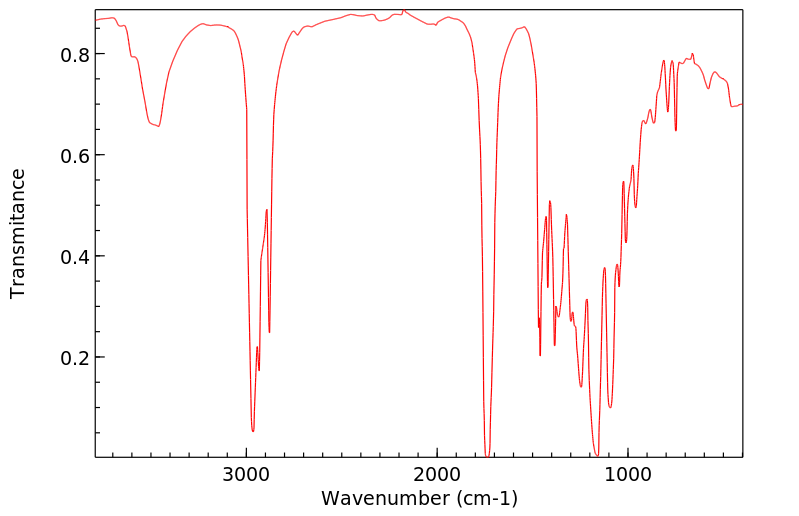
...Tributyrinase.../is/ an enzyme specific for the hydrolysis of tributyrins. This enzyme is inhibited by some organic compounds, particularly selected fluorophosphates. ...A fluoride-sensitive tributyrinase has been isolated from rat adipose tissue, which liberates butyric acid from 1-mono-, 1,2-di-, or tributyrin.
Porcine liver and kidney microsomes most actively hydrolyzed tributyrin. Liver microsomes from frogs, pigs, rats, cats, rabbits, guinea pigs, sheep, doves, dogs but not fish, also possessed esterase activity against tributyrin.
Studies of the hydrolysis of the glycerol fatty acid esters (/including/ tributyrin...) showed complete hydrolysis to glycerol and the corresponding fatty acids, butyric acid, 5-hydroxy-decanoic acid, and 5-hydroxydodecanoic acid, respectively.
Effect of feeding tributyrin on toxicity of diisopropylfluorophosphate (DFP) in mice. After 36 hr of tributyrin feeding the DFP toxicity is 1.7 time normal & after 14 days of feeding it is only 1.15 times normal. .
/HUMAN EXPOSURE STUDIES/ ... We treated 13 patients with escalating doses of tributyrin from 50 to 400 mg/kg/day. Doses were administered p.o. after an overnight fast, once daily for 3 weeks, followed by a 1-week rest. Intrapatient dose escalation occurred after two courses without toxicity greater than grade 2. The time course of butyrate in plasma was assessed on days 1 and 15 and after any dose escalation. Grade 3 toxicities consisted of nausea, vomiting, and myalgia. Grades 1 and 2 toxicities included diarrhea, headache, abdominal cramping, nausea, anemia, constipation, azotemia, lightheadedness, fatigue, rash, alopecia, odor, dysphoria, and clumsiness. There was no consistent increase in hemoglobin F with tributyrin treatment.
/LABORATORY ANIMALS: Acute Exposure/ Inhalation of 78 ppm for 6 hr caused temporary hyperpnea in the rat, but no fatalities or other symptoms were observed.
/LABORATORY ANIMALS: Acute Exposure/ Feeding mice a diet containing 12% tributyrin increased the butyrylcholinesterase activity to about 2.4 times normal & reduced the acetylcholinesterase activity to about 0.55 times normal. These changes intervene between 3rd & 9th day of regimen.
/LABORATORY ANIMALS: Acute Exposure/ ... In Experiment 1, glucose was decreased by intraruminal administration of 84 or 168 g of tributyrin /in goat/. Tributyrin caused transient hyperglycemia immediately after administration. beta-Hydroxybutyrate was increased in a dose-dependent manner by tributyrin, caused large increases of insulin in plasma. In Experiment 2, 84 g of tributyrin administered intraruminally decreased glucose
来源:Hazardous Substances Data Bank (HSDB)
吸收、分配和排泄
Tributyrin在大鼠的肠粘膜被吸收,在豚鼠则不会通过皮肤吸收。
Tributyrin is absorbed by the intestinal mucosa of the rat and is not absorbed cutaneously in guinea pigs.
In humans administered once daily oral injections of 50-400 mg/kg tributyrin for 3 weeks, peak plasma concentrations occurred 0.25-3 hr after dosing. The peak plasma concentration of those administered 200 mg/kg ranged from 0.1-0.45 mM. Higher plasma concentrations were not observed in those given higher doses.
In mice and rats given 10.3 g/kg tributyrin orally, plasma butyrate concentrations peaked at 1.75 and 3.07 mM, respectively. They were >/= 1 mM from 10-60 min after dosing in mice and 30-90 min in rats.
... Female CD2F1 mice were treated with tributyrin by oral gavage or with sodium butyrate by i.v. bolus or oral gavage. Oral tributyrin doses delivered to mice were 3.1, 5.2, 7.8, and 10.3 g/kg. Intravenous sodium butyrate doses were 0.31, 0.62, 0.94, and 1.25 g/kg. Oral sodium butyrate was given to mice at 5 g/kg. Subsequently, similar studies were performed in female Sprague-Dawley rats. Rats were given tributyrin by oral gavage at doses of 3.6, 5.2, or 10.3 g/kg or sodium butyrate i.v. at a dose of 500 mg/kg. Plasma butyrate concentrations were determined by gas chromatography. RESULTS: In mice, oral dosing with tributyrin resulted in detectable plasma butyrate concentrations as early as at 5 min after treatment and produced peak plasma butyrate concentrations at between 15 and 60 min after dosing. Peak plasma butyrate concentrations increased proportionally with increasing tributyrin dose, but as the oral tributyrin dose increased there was a greater than proportional increase in the area under the curve of plasma butyrate concentrations versus time (AUC). At a tributyrin dose of 10.3 g/kg, plasma butyrate concentrations peaked at approximately 1.75 mM and remained >1 mM for between 10 and 60 min after dosing. However, approximately 10% of mice treated with this dose died acutely. At a tributyrin dose of 7.8 g/kg, plasma butyrate concentrations reached approximately 1 mM by 15 min after dosing and remained between 0.8 and 1 mM until 60 min after dosing. No mouse treated with this dose died acutely. Mice given tributyrin doses of 5.2 and 3.1 g/kg achieved peak plasma butyrate concentrations of approximately 0.9 and 0.5 mM, respectively, by 45 min after dosing. Plasma butyrate concentrations in these mice remained above 0.1 mM until 120 and 90 min after dosing, respectively. The four i.v. doses of sodium butyrate resulted in plasma concentration-time profiles that also indicated nonlinear pharmacokinetics and were well described by a one-compartment model with saturable elimination. Values recorded for the Michaelis-Menten constant (Km) and the maximal velocity of the process (Vmax) ranged between 1.02 and 5.65 mM and 0.60 and 1.82 mmol/min, respectively. Values noted for the volume of the central compartment (Vc) varied between 0.48 and 0.72 l/kg. At 1.25 g/kg, i.v. sodium butyrate produced peak plasma butyrate concentrations of 10.5-17.7 mM, and plasma butyrate concentrations remained above 1 mM for 20-30 min. Sodium butyrate delivered orally to mice at 5 g/kg produced peak plasma butyrate concentrations of approximately 9 mM at 15 min after dosing and plasma butyrate concentrations exceeding 1 mM for 90 min after dosing. In rats the 10.3-g/kg oral dose of tributyrin produced peak plasma butyrate concentrations of approximately 3 mM by 75 min after dosing and butyrate concentrations exceeding 1 mM from 30 to 90 min after dosing. The plasma butyrate concentrations produced in rats by 5.2- and 3.6-g/kg doses were appropriately lower than those produced by the 10.3-g/kg dose, and there was no evidence of nonlinearity. The 500-mg/kg i.v. dose of sodium butyrate produced peak plasma butyrate concentrations in rats of approximately 11 mM, and the decline in plasma butyrate concentrations with time after dosing was consistent with saturable clearance.
... Peak plasma butyrate concentrations occurred between 0.25 and 3 h after dose, increased with dose, and ranged from 0 to 0.45 mM. Peak concentrations did not increase in three patients who had dose escalation. Butyrate pharmacokinetics were not different on days 1 and 15. Because peak plasma concentrations near those effective in vitro (0.5-1 mM) were achieved, but butyrate disappeared from plasma by 5 h after dose, we are now pursuing dose escalation with dosing three times daily, beginning at a dose of 450 mg/kg/day.
1.周国泰,化学危险品安全技术全书,化学工业出版社,1997 2.国家环保局有毒化学品管理办公室、北京化工研究院合编,化学品毒性法规环境数据手册,中国环境科学出版社.1992 3.Canadian Centre for Occupational Health and Safety,CHEMINFO Database.1998 4.Canadian Centre for Occupational Health and Safety, RTECS Database, 1989
[EN] COMPOSITIONS AND METHODS FOR THE TREATMENT OF ATHEROTHROMBOSIS<br/>[FR] COMPOSITIONS ET PROCÉDÉS POUR LE TRAITEMENT DE L'ATHÉROTROMBOSE
申请人:KANDULA MAHESH
公开号:WO2013024376A1
公开(公告)日:2013-02-21
The disclosures herein provide compounds of formula I or its pharmaceutical acceptable salts, as well as polymorphs, enantiomers, stereoisomers, solvates, and hydrates thereof. These salts may be formulated as pharmaceutical compositions. The pharmaceutical compositions may be formulated for peroral administration- transdermal administration, transmucosal, syrups, topical, extended release, sustained release, or injection. Such compositions may foe used to treatment of vascular disorders or conditions such as thrombotic cerebrovascular or cardiovascular disease or its associated complications.
[EN] PHOTOCHROMIC AND ELECTROCHROMIC DIARYLETHENE COMPOUNDS WITH IMPROVED PHOTOSTABILITY AND SOLUBILITY<br/>[FR] COMPOSÉS DIARYLÉTHÈNE PHOTOCHROMIQUES ET ÉLECTROCHROMES PRÉSENTANT UNE PHOTOSTABILITÉ ET UNE SOLUBILITÉ AMÉLIORÉES
申请人:SWITCH MAT INC
公开号:WO2020198868A1
公开(公告)日:2020-10-08
A diarylethene compound reversibly convertible under photochromic and electrochromic conditions between a ring-open isomer of Formula (1A) and a ring-closed isomer of Formula (IB) wherein R5 is a substituted phenyl ring and Re is a substituted thiophene ring is provided. The photochromic-electrochromic diarylethene compound of Formula (1A)/(1B) have improved photochromic, electrochromic or photochromic and electrochromic properties, and is useful to provide variation of the light transmission properties of optical filters. The compound also possesses improved solubility making it suitable for incorporation in commercial products..
NOVEL IMMUNOMODULATOR AND ANTI-INFLAMMATORY COMPOUNDS
申请人:MUTHUPPALANIAPPAN Meyyappan
公开号:US20110275603A1
公开(公告)日:2011-11-10
The present invention provides dihydroorotate dehydrogenase inhibitors, methods of preparing them, pharmaceutical compositions containing them and methods of treatment, prevention and/or amelioration of diseases or disorders wherein the inhibition of Dihydroorotate dehydrogenase is known to show beneficial effect.
[EN] (HETERO)ARYL CYCLOPROPYLAMINE COMPOUNDS AS LSD1 INHIBITORS<br/>[FR] COMPOSÉS D'(HÉTÉRO)ARYL-CYCLOPROPYLAMINE À TITRE D'INHIBITEURS DE LSD1
申请人:ORYZON GENOMICS SA
公开号:WO2013057322A1
公开(公告)日:2013-04-25
The invention relates to (hetero)aryl cyclopropylamine compounds, including particularly the compounds of formula (I) as described and defined herein, and their use in therapy, including, e.g., in the treatment or prevention of cancer, a neurological disease or condition, or a viral infection.
[EN] (HETERO)ARYL CYCLOPROPYLAMINE COMPOUNDS AS LSD1 INHIBITORS<br/>[FR] COMPOSÉS (HÉTÉRO)ARYLE CYCLOPROPYLAMINES EN TANT QU'INHIBITEURS DE LSD1
申请人:ORYZON GENOMICS SA
公开号:WO2013057320A1
公开(公告)日:2013-04-25
The invention relates to (hetero)aryl cyclopropylamine compounds, including particularly the compounds of formula I as described and defined herein, and their use in therapy, including, e.g., in the treatment or prevention of cancer, a neurological disease or condition, or a viral infection.