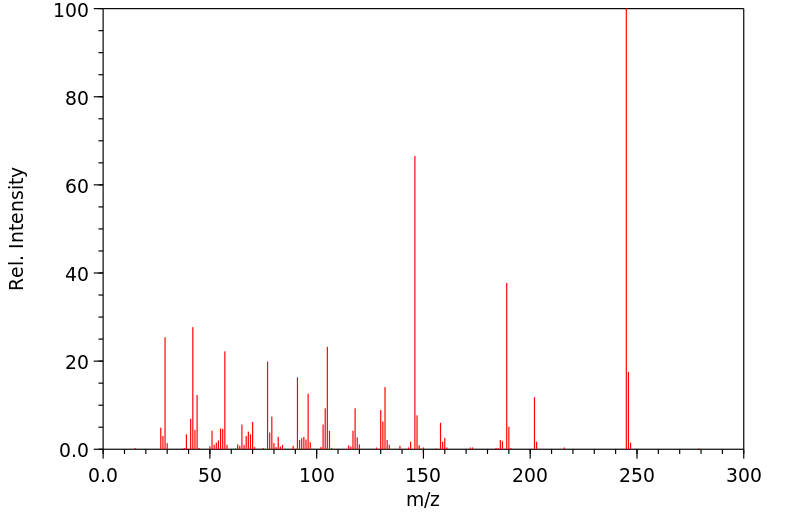
Fentanyl is metabolized to a number of inactive metabolites. Fentanyl is 99% N-dealkylated to norfentanyl by cytochrome P450. It can also be amide hydrolyzed to despropionylfentanyl, or alkyl hydroxylated to hydroxyfentanyl which is N-dealkylated to hydroxynorfentanyl.
Fentanyl does not appear to be metabolized in skin when administered transdermally. Data from clinical studies and from studies using a human keratinocyte cell assay indicate that about 92% of a dose delivered from the fentanyl transdermal system is accounted for as unchanged drug in systemic circulation. Total plasma clearance of fentanyl is reported to be about 500 mL/hour per kg (range: 300-700 mL/hour per kg) or 42-53 L/hour.
Fentanyl citrate is metabolized extensively in the liver and the intestinal mucosa. Animal studies indicate that the drug undergoes oxidation via the microsomal enzymes in the liver and intestinal mucosa (principally cytochrome P-450 [CYP] isoform 3A4) to form norfentanyl; the drug also undergoes hydrolysis to form 4-N-anilinopiperidine and propionic acid. Norfentanyl has been shown to be pharmacologically inactive in animal studies. Fentanyl is excreted in the urine as inactive metabolites and as unchanged drug. Less than 10% of a dose is excreted in urine unchanged and only about 1% is excreted in the feces as unchanged drug.
This study was undertaken to determine if metabolites of fentanyl might be useful in the detection and monitoring of substance abuse. The presence of fentanyl and two of its metabolites in the urine and saliva of seven female patients receiving small doses (110 +/- 56 micrograms) of fentanyl was studied up to 96 hr from the time of administration. Fentanyl and its two metabolites (norfentanyl and despropionylfentanyl) were extracted from samples and analyzed by gas chromatography/mass spectrometry. Unchanged fentanyl was detectable in urine in all patients immediately postoperatively and in 3 of 7 patients at 24 hr. By 72 hr, fentanyl was undetectable. Norfentanyl was present in larger quantities than fentanyl immediately postoperatively and was detected in all patients at 48 hr and in 4 of 7 patients at 96 hr. Despropionylfentanyl was not detected in any of the urine specimens tested. Neither fentanyl nor its metabolites could be detected consistently at any time in saliva. Saliva testing does not appear to be a viable alternative to urine testing based on this study. Urinary norfentanyl might be considered as the substance of choice when testing for fentanyl abuse.
IDENTIFICATION AND USE: Fentanyl is a solid. It is a Schedule II controlled substance. Fentanyl citrate is a strong analgesic used preoperatively, during surgery, and in the immediate postoperative period for its analgesic action. In addition, the drug may be used to prevent or relieve tachypnea and postoperative emergence delirium. HUMAN STUDIES: Introduced into clinical practice in the 1960s, the analgesic fentanyl is 100 times more potent than morphine. Various methods of administration exist including the transdermal patch system, widely used in chronic pain and palliative care settings. Numerous, often imaginative methods of abuse of fentanyl patches have been reported, the majority of fatal fentanyl overdose cases resulting from deliberate abuse or suicide. The most serious adverse effect of fentanyl is respiratory depression. Fentanyl should be administered only under the supervision of qualified clinicians. Patients receiving fentanyl should be advised of the importance of taking the drug exactly as prescribed. Skeletal and thoracic muscle rigidity occurs frequently, especially following rapid IV administration of fentanyl. Muscular rigidity may be associated with reduced pulmonary compliance and/or apnea, laryngospasm, and bronchoconstriction and may be managed by use of assisted or controlled respiration or, if necessary, by IV administration of a neuromuscular blocking agent. Bradycardia may occur following administration of fentanyl and may be controlled with atropine. Delayed chest wall muscle rigidity may develop up to 24 hours after administration or 9 hours after after ending an IV infusion and may persist for hours. Masseter muscle spasm and laryngospasm may also occur. Acute overdosage produces signs of opioid toxicity: circulatory and CNS depression, lethargy, coma, respiratory depression, and decreased GI motility with ileus. Miosis is a variable finding, depending on the particular opioid, co-ingestants, and secondary effects, such as hypoxia. Apnea, hypotension, bradycardia, non-cardiogenic pulmonary edema, seizures, dysrhythmias, and death may occur with severe poisoning. With continued use, tolerance to the pharmacologic and adverse effects of the drug develop, but the rate of development of tolerance exhibits considerable interindividual variation. ANIMAL STUDIES: Fentanyl was not teratogenic in rats at dosages of 10-500 ug/kg daily when administered on days 7-21 of gestation. At an IV dosage of 10-30 ug/kg daily on days 6-18 of gestation in rats, fentanyl was embryotoxic or fetotoxic and slightly increased mean delivery time in animals receiving the highest dosage, but the drug was not teratogenic. At IV doses of 30 ug/kg or subcutaneous doses of 160 ug/kg in rats, fentanyl increased embryonic resorptions. Fentanyl citrate was not mutagenic in the in vitro Ames reverse mutation assay in S. typhimurium or E. coli, or the mouse lymphoma mutagenesis assay, and was not clastogenic in the in vivo mouse micronucleus assay. In veterinary use, overdosage may produce profound respiratory and/or CNS depression in most species. Newborns may be more susceptible to these effects than adult animals. Other toxic effects may include cardiovascular collapse, tremors, reck rigidity, and seizures.
Opiate receptors are coupled with G-protein receptors and function as both positive and negative regulators of synaptic transmission via G-proteins that activate effector proteins. Binding of the opiate stimulates the exchange of GTP for GDP on the G-protein complex. As the effector system is adenylate cyclase and cAMP located at the inner surface of the plasma membrane, opioids decrease intracellular cAMP by inhibiting adenylate cyclase. Subsequently, the release of nociceptive neurotransmitters such as substance P, GABA, dopamine, acetylcholine and noradrenaline is inhibited. Opioids also inhibit the release of vasopressin, somatostatin, insulin and glucagon. Fentanyl's analgesic activity is, most likely, due to its conversion to morphine. Opioids close N-type voltage-operated calcium channels (OP2-receptor agonist) and open calcium-dependent inwardly rectifying potassium channels (OP3 and OP1 receptor agonist). This results in hypopolarization and reduced neuronal excitability.
来源:Toxin and Toxin Target Database (T3DB)
毒理性
致癌物分类
对人类无致癌性(未列入国际癌症研究机构IARC清单)。
No indication of carcinogenicity to humans (not listed by IARC).
来源:Toxin and Toxin Target Database (T3DB)
毒理性
健康影响
医学问题可能包括肺充血、肝病、破伤风、心脏瓣膜感染、皮肤脓肿、贫血和肺炎。过量可能会导致死亡。
Medical problems can include congested lungs, liver disease, tetanus, infection of the heart valves, skin abscesses, anemia and pneumonia. Death can occur from overdose.
◉ Summary of Use during Lactation:When used epidurally or intravenously during labor or for a short time immediately postpartum, amounts of fentanyl ingested by the neonate are usually small and are not expected to cause any adverse effects in breastfed infants. The results of studies on the effect of epidural fentanyl on breastfeeding initiation and duration are mixed, because of the many different combinations of drugs, dosages and patient populations studied as well as the variety of techniques used and deficient designs of many of the studies.
It has been suggested that a cumulative dose of 80 to 150 mcg of fentanyl during labor and delivery reduces breastfeeding success, but another study found no marked decrease in breastfeeding success with doses above 150 mcg in motivated women with previous breastfeeding success.
In infants placed skin-to-skin after a normal vaginal delivery, epidural fentanyl given during labor may delay the infant's first suckling in a dose-dependent manner, perhaps because it can persist in the infant's serum for over 24 hours after discontinuation. However, it appears that with good breastfeeding support, epidural fentanyl plus bupivacaine has little overall effect on breastfeeding success.
No waiting period or discarding of milk is required before resuming breastfeeding after fentanyl is used for short procedures (e.g., for endoscopy). After general anesthesia, breastfeeding can be resumed as soon as the mother has recovered sufficiently from anesthesia to nurse. When a combination of anesthetic agents is used for a procedure, follow the recommendations for the most problematic medication used during the procedure. Limited information indicates that transdermal fentanyl in a dosage of 100 mcg/hour results in undetectable fentanyl concentrations in breastmilk.
Newborn infants seem to be particularly sensitive to the effects of even small dosages of narcotic analgesics. Preterm infants are the most susceptible to fentanyl’s effects because their clearance is substantially reduced. Once the mother's milk comes in, it is best to provide pain control with a nonnarcotic analgesic and limit maternal intake of fentanyl to a few days at a low dosage with close infant monitoring. If the baby shows signs of increased sleepiness (more than usual), difficulty breastfeeding, breathing difficulties, or limpness, a physician should be contacted immediately.
◉ Effects in Breastfed Infants:Fentanyl was possibly the cause of statistically significant, but clinically unimportant, lower neurobehavioral scores in a group of 32 newborns who were less than 24 hours old and whose mothers had received epidural fentanyl during labor.
An epidural fentanyl dosage greater than 150 mcg during labor was associated with slightly lower neurobehavioral scores in the newborns of 177 breastfeeding mothers on postpartum day 1 compared to a lower total dosage or to no fentanyl; however, this might have been a chance association and was probably due to placental transfer of fentanyl prior to delivery and not from breastmilk after delivery. All women also received epidural bupivacaine.
A woman was using a transdermal fentanyl patch for chronic back pain during pregnancy and postpartum. The mother required additional analgesia during labor and the infant required treatment for neonatal abstinence syndrome. By 2 weeks postpartum, the mother was using a fentanyl patch in a dosage of 100 mcg/hour which was changed every other day and the infant was being fed the mother's milk every 3 hours. The infant had no additional medical problems and fed well until discharge after day 27 of life, gaining 500 grams.
A search was performed of the shared database of all U.S. poison control centers for the time period of 2001 to 2017 for calls regarding medications and breastfeeding. Of 2319 calls in which an infant was exposed to a substance via breastmilk, 7 were classified as resulting in a major adverse effect, and one of these involved fentanyl. A one-month-old infant was exposed to fentanyl, morphine, oxycodone, and unspecified benzodiazepines. The infant was admitted to the intensive care unit and described as being agitated and irritable and having tachycardia, confusion, drowsiness, lethargy, miosis, respiratory depression, acidosis, and hyperglycemia. The dosages, routes of administration, and extent of breastfeeding were not reported and the infant survived.
◉ Effects on Lactation and Breastmilk:Fentanyl can increase serum prolactin. However, the prolactin level in a mother with established lactation may not affect her ability to breastfeed.
Five women who were 6 to 15 weeks postpartum were given single doses of 100 mcg of fentanyl, 2 mg of midazolam and 2.5 mg/kg of propofol intravenously before undergoing general anesthesia. The women's milk output following the surgical procedure was less than half of the normal milk output of nursing mothers. The authors speculated that milk volume might be reduced postoperatively because of perioperative fluid restriction and volume losses, as well as stress-induced inhibition of milk production.
In 58 breastfeeding mothers who received an epidural fentanyl dosage greater than 150 mcg during labor, 21% reported more difficulty in establishing breastfeeding at 24 hours after delivery compared to 10% of mothers who received to a lower dosage or to no fentanyl. There was no difference in breastfeeding difficulty noted between the groups 24 hours after delivery as determined by a lactation consultant. Women in the high-dose group who could be contacted were more likely to discontinue breastfeeding by 6 weeks after delivery and there was a higher rate of breastfeeding discontinuation at 6 weeks among mothers who reported breastfeeding difficulty 24 hours after delivery. A relatively high dropout rate from the study at 6 weeks clouds the results.
A retrospective study of a random sample of 425 mothers delivering in a maternity unit found a dose-related increased risk of bottle feeding at hospital discharge associated with fentanyl administered during labor.
A prospective cohort study compared women who received continuous epidural analgesia with fentanyl and either bupivacaine or ropivacaine during labor and delivery (n = 52) to women who received no analgesia (n = 63). The average total fentanyl dosage was 124 mcg and the average total infusion time from start to delivery was 219 minutes. The study found no differences between the groups in breastfeeding effectiveness or infant neurobehavioral status at 8 to 12 hours postpartum or the number exclusively or partially breastfeeding at 4 weeks postpartum.
A randomized, prospective study measured infant breastfeeding behavior following epidural or intravenous fentanyl during delivery in 100 multiparous mothers undergoing cesarean section and delivering fullterm, healthy infants. Epidural fentanyl was given to 50 women in a dose of 100 to 150 mcg in divided doses followed by a continuous epidural infusion of 20 mcg/hour. Intravenous fentanyl was given to 50 women as a single dose of 50 mcg after delivery. Both groups received epidural or spinal bupivacaine in addition. A slight difference was seen in breastfeeding behavior between the groups, with the infants in the intravenous fentanyl group performing slightly worse than those in the epidural group. However, all mothers were able to breastfeed their infants at 24 hours. None had severe breastfeeding problems; 10 women in the epidural group reported mild or moderate problems and 7 women in the intravenous group reported breastfeeding problems. Twenty mothers in the epidural group and 14 in the intravenous group used supplemental bottle feeding, with the difference not statistically significant.
A randomized, multicenter trial compared the initiation rate and duration of breastfeeding in women who received high-dose epidural bupivacaine alone, or one of two low-dose combinations of bupivacaine plus fentanyl. The average fentanyl dosages in the two groups were 97 and 151 mcg in the first stage of labor and 10 and 12 mcg of fentanyl during the second stage of labor, respectively, with great variability. A nonepidural matched control group was also compared. No differences in breastfeeding initiation rates or duration were found among the epidural and nonmedicated groups, but women in the nonepidural group who received systemic meperidine had a lower breastfeeding initiation rate than in the other groups.
A nonrandomized study in low-risk mother-infant pairs found that there was no difference overall in the amount of sucking by newborns, whether their mothers received bupivacaine plus fentanyl, or fentanyl alone by epidural infusion in various dosages, or received no analgesia for childbirth. In a subanalysis by sex and number of sucks, female infants were affected by high-dose bupivacaine and high-dose fentanyl, but male infant were not. However, the imbalances of many factors between the study groups makes this study difficult to interpret.
In a prospective cohort study, 87 multiparous women who received epidural bupivacaine and fentanyl for pain control during labor and vaginal delivery. A loading dose of 0.125% bupivacaine with fentanyl 50-100 mcg. Epidural analgesia is maintained using 0.0625% bupivacaine and fentanyl 0.2 mcg/mL. The median dose of fentanyl received by the women was 151 mcg (range 30 to 570 mcg). The women completed questionnaires at 1 and 6 weeks postpartum regarding breastfeeding. Most women had prior experience with breastfeeding, support at home and ample time off from work. All women were breastfeeding at 1 week postpartum and 95.4% of women were breastfeeding at 6 weeks postpartum.
A nonrandomized study at one Italian hospital compared primiparous mothers undergoing vaginal delivery who received epidural analgesia (n = 64) to those who did not (n = 64). Mothers who requested the epidural analgesia received an initial dose of 100 mcg of fentanyl diluted to 10 mL with saline. After the initial fentanyl, doses of 15 to 20 mL of 0.1% ropivacaine were administered if needed. The only difference between the groups of mothers was a longer duration of labor among the treated mothers. The quality of infant nursing was equal between the 2 groups of infants on several measures; however, more infants in the treated group breastfed for less than 30 minutes at the first feeding.
A national survey of women and their infants from late pregnancy through 12 months postpartum compared the time of lactogenesis II in mothers who did and did not receive pain medication during labor. Categories of medication were spinal or epidural only, spinal or epidural plus another medication, and other pain medication only. Women who received medications from any of the categories had about twice the risk of having delayed lactogenesis II (>72 hours) compared to women who received no labor pain medication.
A randomized study compared the effects of cesarean section using general anesthesia, spinal anesthesia, or epidural anesthesia, to normal vaginal delivery on serum prolactin and oxytocin as well as time to initiation of lactation. General anesthesia was performed using propofol 2 mg/kg and rocuronium 0.6 mg/kg for induction, followed by sevoflurane and rocuronium 0.15 mg/kg as needed. After delivery, patients in all groups received an infusion of oxytocin 30 international units in 1 L of saline, and 0.2 mg of methylergonovine if they were not hypertensive. Fentanyl 1 to 1.5 mcg/kg was administered after delivery to the general anesthesia group. Patients in the general anesthesia group (n = 21) had higher post-procedure prolactin levels and a longer mean time to lactation initiation (25 hours) than in the other groups (10.8 to 11.8 hours). Postpartum oxytocin levels in the nonmedicated vaginal delivery group were higher than in the general and spinal anesthesia groups.
A randomized, nonblinded study compared the use of intramuscular meperidine 100 mg to intranasal (mean dose 486 mcg) or subcutaneous (mean dose 300 mcg) fentanyl for labor analgesia. More women in the meperidine group had difficulty establishing lactation (79%) than in the intranasal (39%) or subcutaneous (44%) fentanyl groups. Mothers who received meperidine reported more sedation, had longer labors, and their infants were more likely to be admitted to the nursery.
A retrospective study in a Spanish public hospital compared the infants of mothers who received an epidural during labor that contained fentanyl and either bupivacaine or ropivacaine. Infants of mothers who received an epidural had a lower frequency of early breastfeeding.
A small prospective study in California compared women who received an epidural infusion of fentanyl and ropivacaine to mothers who did not receive an epidermal during labor. All mothers had normal vaginal deliveries and their infants had 1 uninterrupted hour of skin-to-skin contact immediately postpartum. The study found inverse relationships between the amount of fentanyl and the amount of oxytocin received during labor and the time of the first suckling. Because women who received more fentanyl also tended to receive more oxytocin, the study could not clearly separate the effects of the two drugs.
A randomized, double-blind study compared three epidural maintenance solutions for labor analgesia in women receiving epidural analgesia during labor: bupivacaine 1 mg/mL, bupivacaine 0.8 mg/mL with fentanyl 1 mcg/mL, or bupivacaine 0.625 mg/mL with fentanyl 2 mcg/mL. At 6 weeks postpartum, the breastfeeding rate was 94% or greater in all groups, with no difference among them. All mothers delivered full-term infants and were highly motivated to breastfeed and almost all had vaginal deliveries.
A prospective cohort study in 1204 Israeli women on the effect of labor epidural analgesia during labor, the following protocol was used: bupivacaine 0.1% 15 mL and fentanyl 100 mcg in 5-mL increments, followed by an epidural infusion of bupivacaine 0.1% 10 mL and fentanyl 2 mcg/mL, with a patient-controlled epidural analgesia modality with 5 mL bolus with a lock-out time of 15 minutes. At 6 weeks postpartum, the breastfeeding and exclusive breastfeeding rates were lower (74% and 52%, respectively) in mothers who received the epidural analgesia than in those who did not (83% and 68%, respectively). However, the difference was mostly accounted for by parity, with the intervention having little effect on multiparous women.
A prospective study in an Australian hospital compared mothers who received epidural fentanyl analgesia, subcutaneous morphine or neither during labor and delivery. When controlled for labor induction, instrumental delivery and special care nursery admission, no difference was seen between the 3 groups in breastfeeding rates at discharge or at 6 weeks postpartum.
A randomized, partially blinded study in a hospital in Thailand compared intravenous meperidine and fentanyl for pain during active labor. Mothers received either meperidine 50 mg (n = 46) or fentanyl 50 mcg (n = 46) initially and then every 1 (fentanyl) or 2 (meperidine) hours as requested by the mother. The percentages of infants who breastfed in the first 24 hours were only 61% for meperidine and 54% for fentanyl, although the difference was not statistically significant. Care of the infants (e.g., skin-to-skin in the first hour) was not reported.
A multicenter, prospective cohort study in Hong Kong of 1277 women who gave birth found that women who received epidural analgesia with either fentanyl or morphine had no decreased frequency of breastfeeding in the first hour compared to mothers who did not receive epidural analgesia. All epidural injections were combined with a local anesthetic, but the exact dosages were not given.
A prospective, observational study in Norway found that infant spontaneous suckling was negatively associated with any intrapartum fentanyl use. The odds of non-exclusive breastfeeding were doubled with epidural fentanyl analgesia and were 4 times higher with intravenous plus epidural fentanyl compared to no opioid exposure. Compared to higher doses, intravenous fentanyl doses greater than 200 mcg resulted in a reduction in exclusive breastfeeding and spontaneous suckling, and an increase in breastfeeding problems.
A nonrandomized, nonblinded study in a Serbian hospital of women near term who underwent cesarean section compared general anesthesia (n = 284) to spinal or epidural anesthesia (n = 249). Spinal anesthesia consisted of hyperbaric bupivacaine 12 mg and fentanyl 0.01 mg; epidural anesthesia consisted of isobaric bupivacaine 0.5% (0.5 mg per 10 cm height) and fentanyl 0.05 mg. General anesthesia consisted of propofol 2.3 mg/kg and succinylcholine 1.5 mg/kg for induction and intubation, followed by an anesthetic gas mixture and oxygen. Reportedly, nitric oxide (possibly nitrous oxide) was 50% of the gas before delivery and 67% after delivery. Sevoflurane was also used in some cases. After delivery and cord clamping, mothers received fentanyl 3 mcg/kg and rocuronium 0.5 mg/kg intravenously for placental delivery. After surgery, neuromuscular block reversal was performed with neostigmine and atropine. All patients received 1 mg/kg of diclofenac every 8 h for 24 hours after delivery and 98% of general anesthesia patients also received 100 mg of tramadol and 78.5% received acetaminophen 1 gram. No regional anesthesia patients received tramadol or acetaminophen. Patients receiving one of the regional anesthetic protocols established lactation sooner (56% and 29% after 18 and 24 hours, respectively), while 86% of women receiving general anesthesia did not establish lactation until 36 to 48 hours after surgery.
Fentanyl sublingual tablets are 54% bioavailable, transmucosal lozenges are 50% bioavailable, buccal tablets are 65% bioavailable, sublingual spray is 76% bioavailable, and nasal spray is 20% more bioavailable than transmucosal (or approximately 64% bioavailable). Fentanyl transmucosal lozenges reach a Cmax of 0.4±0.1ng/mL for a 200µg dose and 2.5±0.6ng/mL for a 1600µg dose with a Tmax of 20-40 minutes. The AUC was 172±96ng\*min/mL for a 200µg dose and 1508±1360ng\*min/mL for a 1600µg dose. Fentanyl sublingual spray reached a Cmax of 0.20±0.06ng/mL for a 100µg dose and 1.61±0.60ng/mL for an 800µg dose with a Tmax of 0.69-1.25 hours, decreasing as the dose increased. The AUC was 1.25±0.67ng\*h/mL for a 100µg dose and 10.38±3.70ng\*h/mL for a 800µg dose. Fentanyl transdermal systems reached a Cmax of 0.24±0.20ng/mL with a Tmax of 3.6±1.3h for a 25µg/h dose. The AUC was 0.42±0.35ng/mL\*h. Fentanyl nasal spray reaches a Cmax of 815±301pg/mL with a Tmax of less than 1 hour for a 200µg/100µL dose. The AUC was 3772pg\*h/mL.
The intravenous volume of distribution is 4L/kg (3-8L/kg). The oral volume of distribution is 25.4L/kg. In hepatically impaired patients, the intravenous volume of distribution ranges from 0.8-8L/kg. Fentanyl crosses the blood brain barrier and the placenta.
Total plasma clearance of fentanyl is 0.5L/hr/kg (0.3-0.7L/hr/kg) or 42L/hr. Following an intravenous dose, surgical patients displayed a clearance of 27-75L/h, hepatically impaired patients displayed a clearance of 3-80L/h, and renally impaired patients displayed a clearance of 30-78L/h.
Because release of fentanyl from fentanyl transdermal systems and percutaneous permeability of the drug are temperature dependent, serum fentanyl concentrations could theoretically increase by approximately one-third in patients with a body temperature of 40 °C. Patients who develop a fever while using fentanyl transdermal system should be observed closely for manifestations of opiate toxicity, and dosage of the drug should be adjusted accordingly. Patients should be cautioned to avoid strenuous exertion that leads to increased core body temperature while wearing the transdermal system. Because application of heat over the fentanyl transdermal system increases mean systemic exposure and peak plasma concentrations of the drug by 120 and 61%, respectively, and has resulted in fatal overdosage, patients wearing a fentanyl transdermal system should be advised to avoid exposing the application site or surrounding area to direct external heat sources.
[EN] S-NITROSOMERCAPTO COMPOUNDS AND RELATED DERIVATIVES<br/>[FR] COMPOSÉS DE S-NITROSOMERCAPTO ET DÉRIVÉS APPARENTÉS
申请人:GALLEON PHARMACEUTICALS INC
公开号:WO2009151744A1
公开(公告)日:2009-12-17
The present invention is directed to mercapto-based and S- nitrosomercapto-based SNO compounds and their derivatives, and their use in treating a lack of normal breathing control, including the treatment of apnea and hypoventilation associated with sleep, obesity, certain medicines and other medical conditions.
[EN] HYBRID MU OPIOID RECEPTOR AND NEUROPEPTIDE FF RECEPTOR BINDING MOLECULES, THEIR METHODS OF PREPARATION AND APPLICATIONS IN THERAPEUTIC TREATMENT<br/>[FR] RÉCEPTEUR D'OPIOÏDE MU HYBRIDE ET MOLÉCULES DE LIAISON DE RÉCEPTEUR DE NEUROPEPTIDE FF, LEURS PROCÉDÉS DE PRÉPARATION ET D'APPLICATIONS DANS UN TRAITEMENT THÉRAPEUTIQUE
申请人:CENTRE NAT RECH SCIENT
公开号:WO2019170919A1
公开(公告)日:2019-09-12
The present invention relates to molecules binding the mu opioid receptor (MOR) and the neuropeptide FF receptor (NPFFR) and in particular molecules having a MOR agonist and NPFFR modulatory activity. The present invention relates to pharmaceutical compositions, and in particular useful in the treatment of pain and/or hyperalgesia.
[EN] METHYL OXAZOLE OREXIN RECEPTOR ANTAGONISTS<br/>[FR] MÉTHYLOXAZOLES ANTAGONISTES DU RÉCEPTEUR DE L'OREXINE
申请人:MERCK SHARP & DOHME
公开号:WO2016089721A1
公开(公告)日:2016-06-09
The present invention is directed to methyl oxazole compounds which are antagonists of orexin receptors. The present invention is also directed to uses of the compounds described herein in the potential treatment or prevention of neurological and psychiatric disorders and diseases in which orexin receptors are involved. The present invention is also directed to compositions comprising these compounds. The present invention is also directed to uses of these compositions in the potential prevention or treatment of such diseases in which orexin receptors are involved.
3-Aminocyclopentanecarboxamides as modulators of chemokine receptors
申请人:Xue Chu-Biao
公开号:US20060004018A1
公开(公告)日:2006-01-05
The present invention is directed to compounds of Formula I:
which are modulators of chemokine receptors. The compounds of the invention, and compositions thereof, are useful in the treatment of diseases related to chemokine receptor expression and/or activity.