
对五联苯 | 3073-05-0
中文名称
对五联苯
中文别名
对联五苯;对五联苯,98%
英文名称
penta-p-phenylene
英文别名
p-quinquephenyl;p-quinquiphenyl;p-quinqephenyl;quinquephenyl;[1,1';4',1'';4'',1''';4''',1'''']Quinquephenyl;1,4-bis(4-phenylphenyl)benzene

CAS
3073-05-0
化学式
C30H22
mdl
——
分子量
382.505
InChiKey
OMCUOJTVNIHQTI-UHFFFAOYSA-N
BEILSTEIN
——
EINECS
——
-
物化性质
-
计算性质
-
ADMET
-
安全信息
-
SDS
-
制备方法与用途
-
上下游信息
-
文献信息
-
表征谱图
-
同类化合物
-
相关功能分类
-
相关结构分类
物化性质
-
熔点:386.45°C
-
沸点:444.64°C (rough estimate)
-
密度:1.1333 (estimate)
-
稳定性/保质期:
遵照规定使用和储存,则不会分解。
计算性质
-
辛醇/水分配系数(LogP):8.9
-
重原子数:30
-
可旋转键数:4
-
环数:5.0
-
sp3杂化的碳原子比例:0.0
-
拓扑面积:0
-
氢给体数:0
-
氢受体数:0
安全信息
-
安全说明:S22,S24/25
-
海关编码:2902909090
SDS
对五联苯 修改号码:5
模块 1. 化学品
产品名称: p-Quinquephenyl
修改号码: 5
模块 2. 危险性概述
GHS分类
物理性危害 未分类
健康危害 未分类
环境危害 未分类
GHS标签元素
图标或危害标志 无
信号词 无信号词
危险描述 无
防范说明 无
模块 3. 成分/组成信息
单一物质/混和物 单一物质
化学名(中文名): 对五联苯
百分比: ....
CAS编码: 3073-05-0
分子式: C30H22
模块 4. 急救措施
吸入: 将受害者移到新鲜空气处,保持呼吸通畅,休息。若感不适请求医/就诊。
皮肤接触: 立即去除/脱掉所有被污染的衣物。用水清洗皮肤/淋浴。
若皮肤刺激或发生皮疹:求医/就诊。
眼睛接触: 用水小心清洗几分钟。如果方便,易操作,摘除隐形眼镜。继续清洗。
如果眼睛刺激:求医/就诊。
食入: 若感不适,求医/就诊。漱口。
紧急救助者的防护: 救援者需要穿戴个人防护用品,比如橡胶手套和气密性护目镜。
模块 5. 消防措施
合适的灭火剂: 干粉,泡沫,雾状水,二氧化碳
对五联苯 修改号码:5
模块 5. 消防措施
特定方法: 从上风处灭火,根据周围环境选择合适的灭火方法。
非相关人员应该撤离至安全地方。
周围一旦着火:如果安全,移去可移动容器。
消防员的特殊防护用具: 灭火时,一定要穿戴个人防护用品。
模块 6. 泄漏应急处理
个人防护措施,防护用具, 使用个人防护用品。远离溢出物/泄露处并处在上风处。
紧急措施: 泄露区应该用安全带等圈起来,控制非相关人员进入。
环保措施: 防止进入下水道。
控制和清洗的方法和材料: 清扫收集粉尘,封入密闭容器。注意切勿分散。附着物或收集物应该立即根据合适的
法律法规处置。
模块 7. 操作处置与储存
处理
技术措施: 在通风良好处进行处理。穿戴合适的防护用具。防止粉尘扩散。处理后彻底清洗双手
和脸。
注意事项: 如果粉尘或浮质产生,使用局部排气。
操作处置注意事项: 避免接触皮肤、眼睛和衣物。
贮存
储存条件: 保持容器密闭。存放于凉爽、阴暗处。
远离不相容的材料比如氧化剂存放。
包装材料: 依据法律。
模块 8. 接触控制和个体防护
工程控制: 尽可能安装封闭体系或局部排风系统,操作人员切勿直接接触。同时安装淋浴器和洗
眼器。
个人防护用品
呼吸系统防护: 防尘面具。依据当地和政府法规。
手部防护: 防护手套。
眼睛防护: 安全防护镜。如果情况需要,佩戴面具。
皮肤和身体防护: 防护服。如果情况需要,穿戴防护靴。
模块 9. 理化特性
固体
外形(20°C):
外观: 晶体-粉末
颜色: 白色
气味: 无资料
pH: 无数据资料
熔点: 无资料
沸点/沸程 无资料
闪点: 无资料
爆炸特性
爆炸下限: 无资料
爆炸上限: 无资料
密度: 无资料
溶解度:
[水] 无资料
[其他溶剂] 无资料
对五联苯 修改号码:5
模块 10. 稳定性和反应性
化学稳定性: 一般情况下稳定。
危险反应的可能性: 未报道特殊反应性。
须避免接触的物质 氧化剂
危险的分解产物: 一氧化碳, 二氧化碳
模块 11. 毒理学信息
急性毒性: 无资料
对皮肤腐蚀或刺激: 无资料
对眼睛严重损害或刺激: 无资料
生殖细胞变异原性: 无资料
致癌性:
IARC = 无资料
NTP = 无资料
生殖毒性: 无资料
模块 12. 生态学信息
生态毒性:
鱼类: 无资料
甲壳类: 无资料
藻类: 无资料
残留性 / 降解性: 无资料
潜在生物累积 (BCF): 无资料
土壤中移动性
log水分配系数: 无资料
土壤吸收系数 (Koc): 无资料
亨利定律 无资料
constaNT(PaM3/mol):
模块 13. 废弃处置
如果可能,回收处理。请咨询当地管理部门。建议在可燃溶剂中溶解混合,在装有后燃和洗涤装置的化学焚烧炉中
焚烧。废弃处置时请遵守国家、地区和当地的所有法规。
模块 14. 运输信息
联合国分类: 与联合国分类标准不一致
UN编号: 未列明
模块 15. 法规信息
《危险化学品安全管理条例》(2002年1月26日国务院发布,2011年2月16日修订): 针对危险化学品的安全使用、
生产、储存、运输、装卸等方面均作了相应的规定。
对五联苯 修改号码:5
模块16 - 其他信息
N/A
模块 1. 化学品
产品名称: p-Quinquephenyl
修改号码: 5
模块 2. 危险性概述
GHS分类
物理性危害 未分类
健康危害 未分类
环境危害 未分类
GHS标签元素
图标或危害标志 无
信号词 无信号词
危险描述 无
防范说明 无
模块 3. 成分/组成信息
单一物质/混和物 单一物质
化学名(中文名): 对五联苯
百分比: ....
CAS编码: 3073-05-0
分子式: C30H22
模块 4. 急救措施
吸入: 将受害者移到新鲜空气处,保持呼吸通畅,休息。若感不适请求医/就诊。
皮肤接触: 立即去除/脱掉所有被污染的衣物。用水清洗皮肤/淋浴。
若皮肤刺激或发生皮疹:求医/就诊。
眼睛接触: 用水小心清洗几分钟。如果方便,易操作,摘除隐形眼镜。继续清洗。
如果眼睛刺激:求医/就诊。
食入: 若感不适,求医/就诊。漱口。
紧急救助者的防护: 救援者需要穿戴个人防护用品,比如橡胶手套和气密性护目镜。
模块 5. 消防措施
合适的灭火剂: 干粉,泡沫,雾状水,二氧化碳
对五联苯 修改号码:5
模块 5. 消防措施
特定方法: 从上风处灭火,根据周围环境选择合适的灭火方法。
非相关人员应该撤离至安全地方。
周围一旦着火:如果安全,移去可移动容器。
消防员的特殊防护用具: 灭火时,一定要穿戴个人防护用品。
模块 6. 泄漏应急处理
个人防护措施,防护用具, 使用个人防护用品。远离溢出物/泄露处并处在上风处。
紧急措施: 泄露区应该用安全带等圈起来,控制非相关人员进入。
环保措施: 防止进入下水道。
控制和清洗的方法和材料: 清扫收集粉尘,封入密闭容器。注意切勿分散。附着物或收集物应该立即根据合适的
法律法规处置。
模块 7. 操作处置与储存
处理
技术措施: 在通风良好处进行处理。穿戴合适的防护用具。防止粉尘扩散。处理后彻底清洗双手
和脸。
注意事项: 如果粉尘或浮质产生,使用局部排气。
操作处置注意事项: 避免接触皮肤、眼睛和衣物。
贮存
储存条件: 保持容器密闭。存放于凉爽、阴暗处。
远离不相容的材料比如氧化剂存放。
包装材料: 依据法律。
模块 8. 接触控制和个体防护
工程控制: 尽可能安装封闭体系或局部排风系统,操作人员切勿直接接触。同时安装淋浴器和洗
眼器。
个人防护用品
呼吸系统防护: 防尘面具。依据当地和政府法规。
手部防护: 防护手套。
眼睛防护: 安全防护镜。如果情况需要,佩戴面具。
皮肤和身体防护: 防护服。如果情况需要,穿戴防护靴。
模块 9. 理化特性
固体
外形(20°C):
外观: 晶体-粉末
颜色: 白色
气味: 无资料
pH: 无数据资料
熔点: 无资料
沸点/沸程 无资料
闪点: 无资料
爆炸特性
爆炸下限: 无资料
爆炸上限: 无资料
密度: 无资料
溶解度:
[水] 无资料
[其他溶剂] 无资料
对五联苯 修改号码:5
模块 10. 稳定性和反应性
化学稳定性: 一般情况下稳定。
危险反应的可能性: 未报道特殊反应性。
须避免接触的物质 氧化剂
危险的分解产物: 一氧化碳, 二氧化碳
模块 11. 毒理学信息
急性毒性: 无资料
对皮肤腐蚀或刺激: 无资料
对眼睛严重损害或刺激: 无资料
生殖细胞变异原性: 无资料
致癌性:
IARC = 无资料
NTP = 无资料
生殖毒性: 无资料
模块 12. 生态学信息
生态毒性:
鱼类: 无资料
甲壳类: 无资料
藻类: 无资料
残留性 / 降解性: 无资料
潜在生物累积 (BCF): 无资料
土壤中移动性
log水分配系数: 无资料
土壤吸收系数 (Koc): 无资料
亨利定律 无资料
constaNT(PaM3/mol):
模块 13. 废弃处置
如果可能,回收处理。请咨询当地管理部门。建议在可燃溶剂中溶解混合,在装有后燃和洗涤装置的化学焚烧炉中
焚烧。废弃处置时请遵守国家、地区和当地的所有法规。
模块 14. 运输信息
联合国分类: 与联合国分类标准不一致
UN编号: 未列明
模块 15. 法规信息
《危险化学品安全管理条例》(2002年1月26日国务院发布,2011年2月16日修订): 针对危险化学品的安全使用、
生产、储存、运输、装卸等方面均作了相应的规定。
对五联苯 修改号码:5
模块16 - 其他信息
N/A
上下游信息
-
上游原料
中文名称 英文名称 CAS号 化学式 分子量 对三联苯 [1,1';4',1'']terphenyl 92-94-4 C18H14 230.309 4,4''-二碘对三联苯 4,4''-diiodo-1,1':4',1''-terphenyl 19053-14-6 C18H12I2 482.102 4-碘联苯 4-iodo-biphenyl 1591-31-7 C12H9I 280.108 4-溴代联苯 4-bromo-1,1'-biphenyl 92-66-0 C12H9Br 233.107
反应信息
-
作为反应物:参考文献:名称:对五苯与碱金属化学还原时电荷和芳香度的相互作用摘要:对包含五个对位连接的芳环的对亚苯基(即对五苯(C 30 H 22 , 1))与碱金属在 THF 中的化学还原研究表明,很容易形成双重还原阴离子1 2–,并结晶与不同的碱金属抗衡离子。使用单晶 X 射线衍射和光谱方法对几种产品进行了表征。使用不同的碱金属可以调节所得结晶产物中的金属结合。电子加成和金属络合对p核心的影响借助计算方法对五苯进行了研究。最值得注意的是,还原导致1 2-从局部芳香族特征转变为醌型特征,这种转变通过与碱金属阳离子的络合而减轻。DOI:10.1021/acs.organomet.2c00583
-
作为产物:参考文献:名称:Buu-Hoi; Cagniant, Comptes Rendus Hebdomadaires des Seances de l'Academie des Sciences, 1943, vol. 216, p. 381摘要:DOI:
文献信息
-
Two new treefrogs of the<i>Boophis rappiodes</i>group from eastern Madagascar (Amphibia Mantellidae)作者:M. Vences、F. GlawDOI:10.1080/03946975.2002.10531171日期:2002.8Two new sibling species of Boophis Tschudi 1838 are described from Andasibe in central-eastern Madagascar. Both are small greenish treefrogs with a translucent ventral skin and without lateral fringes along lower arm and tarsus, and are thereby assignable to the phenetic B. rappiodes group. Boophis bottae n. sp. is morphologically similar to B. rappiodes (Ahl 1928) and occurs syntopically with this在马达加斯加中东部的安达西贝描述了两个新的同胞物种Boophis Tschudi 1838。两者都是带有半透明腹侧皮肤的绿色小树蛙,并且在下臂和骨上没有侧向条纹,因此可以归为表象双歧杆菌。Boophis bottae n。sp。在形态上类似于双歧双歧杆菌(Ahl 1928),并且与该物种同位发生。它与同胞的区别很大,其区别在于广告呼叫(长颤音音符而不是两脉冲音符),以及红棕色背侧图案在乙醇中不会很快消失,通常会覆盖整个背部(与红色图案在很大程度上消失了)在双歧杆菌中的乙醇中)。塔布米。sp。类似于红杆菌(B. erythrodactylus)(Guibé1953),但广告呼叫不同(音符由两个而不是四个七个脉冲组成),并且手指和脚趾尖上没有红色。一个选型被指定为红细菌。除了修订的食虫双歧杆菌(B. rapthodeactylus)和B. viridis的分布信息外,还发现了两个新物种Blom
-
Synthesis of cyclopentadienyl iron complexes with substituted phenylene ligands via Suzuki coupling作者:Andrei M. Shved、Yulia V. Nelyubina、Dmitry S. PerekalinDOI:10.1016/j.jorganchem.2019.121061日期:2020.1Cationic cyclopentadienyl iron complexes of substituted phenylenes [CpFe(p-CH3-C6H4-Ar)]PF6 and [CpFe(p-Ar-C6H4-Ar)]PF6 were obtained in 37–73% yields by Pd-catalyzed Suzuki coupling reactions of [CpFe(p-CH3-C6H4-Cl)]PF6 and [CpFe(p-Cl-C6H4-Cl)]PF6 with polyarylboronic acids Ar-B(OH)2 (Ar = biphenyl, naphthyl, phenanthryl, bithienyl). The complex [CpFe(p-CH3-C6H4-Cl)]PF6 also underwent Sonogashira-type取代亚苯基[CpFe(p -CH 3 -C 6 H 4 -Ar)] PF 6和[CpFe(p -Ar-C 6 H 4 -Ar)] PF 6的阳离子环戊二烯基铁络合物的含量为37-73%通过钯催化的[CpFe(p -CH 3 -C 6 H 4 -Cl)] PF 6和[CpFe(p -Cl-C 6 H 4 -Cl)] PF 6与聚芳基硼酸Ar-的Suzuki偶联反应产生乙(OH)2(Ar =联苯,萘基,菲基,联苯二基)。络合物[CpFe的量(p -CH 3 -C 6 H ^ 4 -Cl)] PF 6也经历Sonogashira类与苯乙炔偶联得到化合物[CpFe的量(对- CH 3 -C 6 H ^ 4 -C ≡C-PH ] PF 6的收率为26%。这种偶联反应代表了[CpFe(arene)] PF 6络合物的另一种方法,该络合物通常是在苛刻条件下通过Bolesova-Nesmeyanov反应从二茂铁中获得的(AlCl
-
One Pot Synthesis of<i>p</i>-Polyphenyls<i>via</i>the lntramolecular Cyclization of 3-Dimethyl-aminohex-5-en-1-ynes作者:Marilyn R. Unroe、Bruce A. ReinhardtDOI:10.1055/s-1987-28142日期:——Para linked polyphenyls of various molecular weights are conveniently synthesized from p-bis(3-dimethylamino-1-propynyl)arenes and 1-aryl-2-propenyl bromides in moderate yields using a three step, one-pot procedure.
-
Synthesis of penta-p-phenylenes with oligo(ethylene glycol) side chains作者:J. Manuel López-Romero、Rodrigo Rico、Rocío Martínez-Mallorquín、Jesús Hierrezuelo、Elena Guillén、Chengzhi Cai、J. Carlos Otero、Isabel López-TocónDOI:10.1016/j.tetlet.2007.06.167日期:2007.8We report an efficient synthesis of a series of penta-p-phenylene derivatives with four side chains of various lengths, including deca(ethylene glycol) groups. The key feature of the synthesis is that the side chains are efficiently introduced in the last step, facilitating optimization of the side chains for different applications. Raman spectroscopy study indicates a similarly high rigidity for all
-
Calculated and Experimental UV and IR Spectra of Oligo-para-phenylenes作者:Kwangyong Park、Tae-Won Lee、Min-Ju Yoon、Jong-In ChoeDOI:10.5012/bkcs.2014.35.2.531日期:2014.2.20The quantum mechanical properties of a series of oligo-para-phenylenes (2-11) were characterized using DFT B3LYP/6-311G(d,p) calculations. The global minimum among the various torsional conformers of an oligo-p-phenylene is calculated to be a twist conformation. A less stable planar conformation, in which all the dihedral angles in oligo-p-phenylene are restricted to be planar, has also been calculated. The total electronic energies, normal vibrational modes, Gibbs free energies, and HOMOs and LUMOs of the two different conformations (twisted and planar) of the oligo-p-phenylenes were analyzed. The energy differences between the HOMOs and LUMOs of the substrates are in accord with the maximum absorption peaks of the experimental UV spectra of 2-6. The calculated normal vibrational modes of 2-6 were comparable with their experimental IR spectra.
表征谱图
-
氢谱1HNMR
-
质谱MS
-
碳谱13CNMR
-
红外IR
-
拉曼Raman
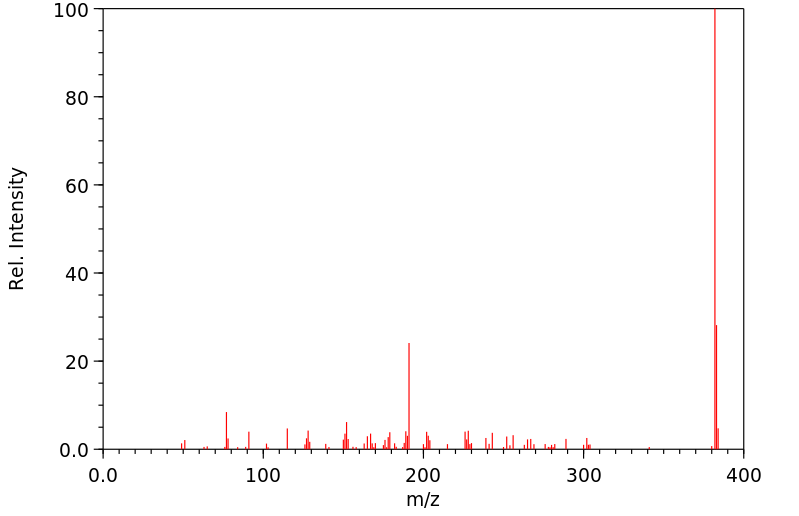
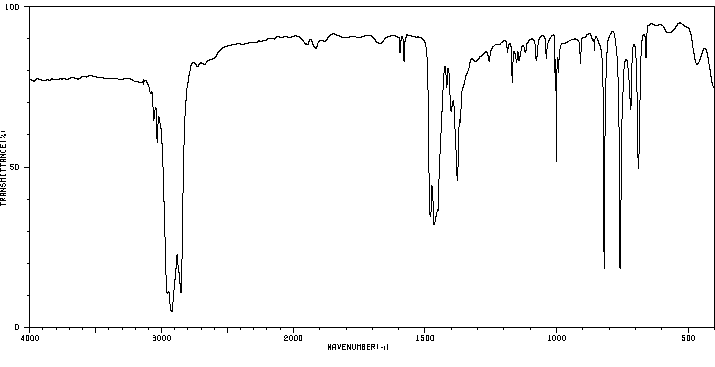
-
峰位数据
-
峰位匹配
-
表征信息
同类化合物
(βS)-β-氨基-4-(4-羟基苯氧基)-3,5-二碘苯甲丙醇
(S,S)-邻甲苯基-DIPAMP
(S)-(-)-7'-〔4(S)-(苄基)恶唑-2-基]-7-二(3,5-二-叔丁基苯基)膦基-2,2',3,3'-四氢-1,1-螺二氢茚
(S)-盐酸沙丁胺醇
(S)-3-(叔丁基)-4-(2,6-二甲氧基苯基)-2,3-二氢苯并[d][1,3]氧磷杂环戊二烯
(S)-2,2'-双[双(3,5-三氟甲基苯基)膦基]-4,4',6,6'-四甲氧基联苯
(S)-1-[3,5-双(三氟甲基)苯基]-3-[1-(二甲基氨基)-3-甲基丁烷-2-基]硫脲
(R)富马酸托特罗定
(R)-(-)-盐酸尼古地平
(R)-(-)-4,12-双(二苯基膦基)[2.2]对环芳烷(1,5环辛二烯)铑(I)四氟硼酸盐
(R)-(+)-7-双(3,5-二叔丁基苯基)膦基7''-[((6-甲基吡啶-2-基甲基)氨基]-2,2'',3,3''-四氢-1,1''-螺双茚满
(R)-(+)-7-双(3,5-二叔丁基苯基)膦基7''-[(4-叔丁基吡啶-2-基甲基)氨基]-2,2'',3,3''-四氢-1,1''-螺双茚满
(R)-(+)-7-双(3,5-二叔丁基苯基)膦基7''-[(3-甲基吡啶-2-基甲基)氨基]-2,2'',3,3''-四氢-1,1''-螺双茚满
(R)-(+)-4,7-双(3,5-二-叔丁基苯基)膦基-7“-[(吡啶-2-基甲基)氨基]-2,2”,3,3'-四氢1,1'-螺二茚满
(R)-3-(叔丁基)-4-(2,6-二苯氧基苯基)-2,3-二氢苯并[d][1,3]氧杂磷杂环戊烯
(R)-2-[((二苯基膦基)甲基]吡咯烷
(R)-1-[3,5-双(三氟甲基)苯基]-3-[1-(二甲基氨基)-3-甲基丁烷-2-基]硫脲
(N-(4-甲氧基苯基)-N-甲基-3-(1-哌啶基)丙-2-烯酰胺)
(5-溴-2-羟基苯基)-4-氯苯甲酮
(5-溴-2-氯苯基)(4-羟基苯基)甲酮
(5-氧代-3-苯基-2,5-二氢-1,2,3,4-oxatriazol-3-鎓)
(4S,5R)-4-甲基-5-苯基-1,2,3-氧代噻唑烷-2,2-二氧化物-3-羧酸叔丁酯
(4S,4''S)-2,2''-亚环戊基双[4,5-二氢-4-(苯甲基)恶唑]
(4-溴苯基)-[2-氟-4-[6-[甲基(丙-2-烯基)氨基]己氧基]苯基]甲酮
(4-丁氧基苯甲基)三苯基溴化磷
(3aR,8aR)-(-)-4,4,8,8-四(3,5-二甲基苯基)四氢-2,2-二甲基-6-苯基-1,3-二氧戊环[4,5-e]二恶唑磷
(3aR,6aS)-5-氧代六氢环戊基[c]吡咯-2(1H)-羧酸酯
(2Z)-3-[[(4-氯苯基)氨基]-2-氰基丙烯酸乙酯
(2S,3S,5S)-5-(叔丁氧基甲酰氨基)-2-(N-5-噻唑基-甲氧羰基)氨基-1,6-二苯基-3-羟基己烷
(2S,2''S,3S,3''S)-3,3''-二叔丁基-4,4''-双(2,6-二甲氧基苯基)-2,2'',3,3''-四氢-2,2''-联苯并[d][1,3]氧杂磷杂戊环
(2S)-(-)-2-{[[[[3,5-双(氟代甲基)苯基]氨基]硫代甲基]氨基}-N-(二苯基甲基)-N,3,3-三甲基丁酰胺
(2S)-2-[[[[[((1S,2S)-2-氨基环己基]氨基]硫代甲基]氨基]-N-(二苯甲基)-N,3,3-三甲基丁酰胺
(2S)-2-[[[[[[((1R,2R)-2-氨基环己基]氨基]硫代甲基]氨基]-N-(二苯甲基)-N,3,3-三甲基丁酰胺
(2-硝基苯基)磷酸三酰胺
(2,6-二氯苯基)乙酰氯
(2,3-二甲氧基-5-甲基苯基)硼酸
(1S,2S,3S,5S)-5-叠氮基-3-(苯基甲氧基)-2-[(苯基甲氧基)甲基]环戊醇
(1S,2S,3R,5R)-2-(苄氧基)甲基-6-氧杂双环[3.1.0]己-3-醇
(1-(4-氟苯基)环丙基)甲胺盐酸盐
(1-(3-溴苯基)环丁基)甲胺盐酸盐
(1-(2-氯苯基)环丁基)甲胺盐酸盐
(1-(2-氟苯基)环丙基)甲胺盐酸盐
(1-(2,6-二氟苯基)环丙基)甲胺盐酸盐
(-)-去甲基西布曲明
龙蒿油
龙胆酸钠
龙胆酸叔丁酯
龙胆酸
龙胆紫-d6
龙胆紫

联系我们



关注我们

公众号

