
5-cyano-6-(4-chlorophenyl)-2-thiouracil | 70638-56-1
中文名称
——
中文别名
——
英文名称
5-cyano-6-(4-chlorophenyl)-2-thiouracil
英文别名
6-(4-chlorophenyl)-4-oxo-2-thioxo-1,2,3,4-tetrahydropyrimidine-5-carbonitrile;6-(4-chlorophenyl)-5-cyano-2-thiouracil;6-(4-chlorophenyl)-4-oxo-2-sulfanylidene-1H-pyrimidine-5-carbonitrile

CAS
70638-56-1
化学式
C11H6ClN3OS
mdl
MFCD06762643
分子量
263.707
InChiKey
XHPUHYGYTQUAMV-UHFFFAOYSA-N
BEILSTEIN
——
EINECS
——
-
物化性质
-
计算性质
-
ADMET
-
安全信息
-
SDS
-
制备方法与用途
-
上下游信息
-
文献信息
-
表征谱图
-
同类化合物
-
相关功能分类
-
相关结构分类
物化性质
-
熔点:252 °C
-
密度:1.55±0.1 g/cm3(Predicted)
计算性质
-
辛醇/水分配系数(LogP):1.8
-
重原子数:17
-
可旋转键数:1
-
环数:2.0
-
sp3杂化的碳原子比例:0.0
-
拓扑面积:97
-
氢给体数:2
-
氢受体数:3
SDS
上下游信息
-
下游产品
中文名称 英文名称 CAS号 化学式 分子量 —— 4-(4-chlorophenyl)-2-hydrazinyl-6-oxo-1,6-dihydropyrimidine-5-carbonitrile —— C11H8ClN5O 261.67 —— 6-(4-chlorophenyl)-3,4-dihydro-2-methylthio-4-oxopyrimidine-5-carbonitrile 128061-80-3 C12H8ClN3OS 277.734 —— 4-(4-chlorophenyl)-6-oxo-2-propan-2-ylsulfanyl-1H-pyrimidine-5-carbonitrile —— C14H12ClN3OS 305.788 —— 2-Benzylsulfanyl-4-(4-chlorophenyl)-6-hydroxy-pyrimidine-5-carbonitrile —— C18H12ClN3OS 353.832 —— 2-(4-Chloroanilino)-4-(4-chlorophenyl)-6-hydroxy-pyrimidine-5-carbonitrile 273920-47-1 C17H10Cl2N4O 357.199 —— 2-(phenethylthio)-4-(4-chlorophenyl)-6-oxo-1,6-dihydropyrimidine-5-carbonitrile 322390-83-0 C19H14ClN3OS 367.859 —— ethyl 2-((4-(4-chlorophenyl)-5-cyano-6-oxo-1,6-dihydropyrimidin-2-yl)thio)acetate —— C15H12ClN3O3S 349.798 —— 5-(4-chlorophenyl)-7-oxo-1,7-dihydro-[1,2,4]triazolo[4,3-a]-pyrimidine-6-carbonitrile 1393647-09-0 C12H6ClN5O 271.666 —— 2-(4-nitrobenzylthio)-4-(4-chlorophenyl)-6-oxo-1,6-dihydropyrimidine-5-carbonitrile 1203458-15-4 C18H11ClN4O3S 398.829 —— 6-(4-chlorophenyl)-3,4-dihydro-3-methyl-2-methylthio-4-oxopyrimidine-5-carbonitrile 109552-71-8 C13H10ClN3OS 291.761 —— 2-Benzylsulfanyl-4-(4-chlorophenyl)-1-methyl-6-oxopyrimidine-5-carbonitrile 1425974-25-9 C19H14ClN3OS 367.859 —— 2-[[4-(4-chlorophenyl)-5-cyano-6-oxo-1H-pyrimidin-2-yl]sulfanyl]-N-[4-(methylsulfamoyl)phenyl]acetamide 1437691-23-0 C20H16ClN5O4S2 489.963 —— 4-(4-Chloro-phenyl)-1-ethyl-2-ethylsulfanyl-6-oxo-1,6-dihydro-pyrimidine-5-carbonitrile 109532-77-6 C15H14ClN3OS 319.815 —— 2-[[4-(4-chlorophenyl)-5-cyano-6-oxo-1H-pyrimidin-2-yl]sulfanyl]-N-(4-sulfamoylphenyl)propanamide 1437691-38-7 C20H16ClN5O4S2 489.963 —— 7-(4-chlorophenyl)-2,3-dihydro-5-oxo-5H-thiazolo<3,2-a>pyrimidine-6-carbonitrile 109532-69-6 C13H8ClN3OS 289.745 —— 2-Benzylsulfanyl-4-(4-chlorophenyl)-1-ethyl-6-oxopyrimidine-5-carbonitrile 1425974-26-0 C20H16ClN3OS 381.886 —— 2-[[4-(4-chlorophenyl)-5-cyano-6-oxo-1H-pyrimidin-2-yl]sulfanyl]-N-(4-sulfamoylphenyl)butanamide 1437691-48-9 C21H18ClN5O4S2 503.99 —— 8-(4-chlorophenyl)-3,4-dihydro-6-oxo-5H-thiazolo<3,2-a>pyrimidine-7-carbonitrile 109532-70-9 C14H10ClN3OS 303.772 —— 4-chloro-6-(4-chlorophenyl)-2-(methylthio)pyrimidine-5-carbonitrile —— C12H7Cl2N3S 296.18 —— 1-Benzyl-2-benzylsulfanyl-4-(4-chloro-phenyl)-6-oxo-1,6-dihydro-pyrimidine-5-carbonitrile 109532-79-8 C25H18ClN3OS 443.956 - 1
- 2
反应信息
-
作为反应物:参考文献:名称:Novel Pyrimidinone Derivatives: Synthesis, Antitumor and Antimicrobial Evaluation摘要:以6-芳基-4-氧代-2-硫代-1,2,3,4-四氢嘧啶-5-碳腈(4a–d)为起始材料,合成了一系列单烷基和双烷基衍生物5a–j和6a,b。对4a、b、d和5d进行肼解反应,得到肼基衍生物7a–c,随后通过与甲酸和氯乙酰氯反应环化,分别得到三唑嘧啶酮8a–c和嘧啶三嗪酮9a–c。大多数新合成的化合物经过体外抗肿瘤活性评估。化合物6a和b在对白血病、非小细胞肺癌、黑色素瘤和肾癌方面表现出良好的抗癌活性。另一方面,所有合成的化合物均经过体外抗菌和抗真菌活性筛选。化合物5h和j对金黄色葡萄球菌表现出显著活性,而化合物5e、7c和8c对白色念珠菌表现出中等抑制活性。DOI:10.1248/cpb.60.521
-
作为产物:描述:Potassium; 6-(4-chloro-phenyl)-5-cyano-4-oxo-1,4-dihydro-pyrimidine-2-thiolate 在 溶剂黄146 作用下, 以 水 为溶剂, 生成 5-cyano-6-(4-chlorophenyl)-2-thiouracil参考文献:名称:Glycosylation of 2-Thiouracil Derivatives. A Synthetic Approach to 3-Glycosyl-2, 4-dioxypyrimidines摘要:Reaction of 6-aryl-5-cyano-2-thiouracils 2a-d with glycosyl halides 4a,b under alkaline conditions gave the respective bisglycosylated derivatives 5a-h. However, their deacetylation with ammonia in methanol caused a cleavage of the S-glycosyl residue and gave the N-3 glycosylated analogues 6a-h.DOI:10.1080/07328319708001360
文献信息
-
Synthesis of 4-(thiazol-2-ylamino)-benzenesulfonamides with carbonic anhydrase I, II and IX inhibitory activity and cytotoxic effects against breast cancer cell lines作者:Nagwa M. Abdel Gawad、Noha H. Amin、Mohammed T. Elsaadi、Fatma M.M. Mohamed、Andrea Angeli、Viviana De Luca、Clemente Capasso、Claudiu T. SupuranDOI:10.1016/j.bmc.2016.05.016日期:2016.7activity on human breast cancer cell line MCF-7. Human (h) CA isoforms I, II and IX were included in the study. The new sulfonamides showed excellent inhibition of all three isoforms, with KIs in the range of 0.84–702 nM against hCA I, of 0.41–288 nM against hCA II and of 5.6–29.2 against the tumor-associated hCA IX, a validated anti-tumor target, with a sulfonamide (SLC-0111) in Phase I clinical trials
-
Design, synthesis, stereochemical determination, molecular docking study, in silico pre-ADMET prediction and anti-proliferative activities of indole-pyrimidine derivatives as Mcl-1 inhibitors作者:Phoebe F. Lamie、John N. PhiloppesDOI:10.1016/j.bioorg.2021.105335日期:2021.11In this study, fourteen novel indole-pyrimidine hybrids were designed and synthesized. Their chemical structures were confirmed using different spectroscopic techniques (1H NMR, 13C NMR, IR and mass). Their (E) stereochemical configuration was determined theoretically (MM2 property) and experimentally using 2D NMR technique (NOESY experiment). The prepared compounds were subjected to preliminary biological在这项研究中,设计并合成了 14 种新型吲哚-嘧啶杂化物。使用不同的光谱技术(1 H NMR、13 C NMR、IR 和质量)确认了它们的化学结构。它们的 ( E ) 立体化学构型是通过理论(MM2 特性)和实验使用 2D NMR 技术(NOESY 实验)确定的。将制备的化合物作为 Mcl-1 抑制剂进行初步生物学研究。大多数化合物表现出良好的靶向 Mcl-1 蛋白的能力,尤其是7d、7e、7i和7k (K i = 11.19–15.21 nM)。针对 Bcl-XL 和 Bcl-2 蛋白进一步评估了这些衍生物。发现一些化合物具有双重 Mcl-1/Bcl-XL 如7i或 Bcl-XL/Bcl-2 抑制活性如7d。 选择最有效的衍生物作为 Mcl-1 抑制剂作为确定对 PC-3、K-562 和 MDA-MB-231 细胞系的体外抗增殖活性的代表性实例。它们具有极好的抗增殖活性。所有合成的化合物都停靠在
-
Part III: Novel checkpoint kinase 2 (Chk2) inhibitors; design, synthesis and biological evaluation of pyrimidine-benzimidazole conjugates作者:Shadia A. Galal、Muhammad Khattab、Samia A. Shouman、Raghda Ramadan、Omaima M. Kandil、Omnia M. Kandil、Ashraf Tabll、Yasmine S. El Abd、Reem El-Shenawy、Yasmin M. Attia、Ahmed A. El-Rashedy、Hoda I. El DiwaniDOI:10.1016/j.ejmech.2018.01.072日期:2018.2cancer therapy. Checkpoint kinase 2 (Chk2) inhibitors offer a promising approach to enhance the effectiveness of cancer chemotherapy. Accordingly, in this study many pyrimidine-benzimidazole conjugates were designed and twelve feasible derivatives were selected to be synthesized to investigate their activity against Chk2 and subjected to study their antitumor activity alone and in combination with the最近,在靶向癌症治疗领域已显示出癌症药物发现的显着发展。Checkpoint激酶2(Chk2)抑制剂为增强癌症化学疗法的有效性提供了一种有前途的方法。因此,在这项研究中,设计了许多嘧啶-苯并咪唑共轭物,并选择了十二种可行的衍生物进行合成,以研究其对Chk2的活性,并单独研究其抗癌活性,并与遗传毒性抗癌药顺铂和阿霉素联用,对乳腺癌进行研究, (ER +)细胞系(MCF-7)。结果表明,所研究的化合物以高效力抑制了Chk2活性(IC 50 = 5.56 nM-46.20 nM)。被研究的候选人对MCF-7(IG 50)表现出显着的抗肿瘤活性 = 6.6μM-24.9μM)。化合物10a-c,14和15显着增强了所研究的遗传毒性药物的活性,而化合物9b和20-23则拮抗了它们的活性。此外,化合物10b与顺铂的组合显示出最佳的凋亡作用,以及化合物10b与阿霉素的组合导致S期的细胞周期完全停滞,其中超过40%的细胞处于S期,而在G2
-
Synthesis and Fungicidal Activity of New Imidazoles from 2-(Chloromethyl)-1<i>H</i>-benzimidazole作者:H. M. F. Madkour、A. A. Farag、S. Sh. Ramses、N. A. A. IbrahiemDOI:10.1080/104265090970241日期:2006.2A series of substituted 2-thiomethylbenzimidazoles 2–4, 2-phenoxy-methylbenzimidazoles 5 and 2-aminomethylbenzimidazoles 6 and 7 were synthesized by reactions of 2-chloromethylbenzimidazole 1 with dithiocarbamate, pyrimidine-2-thiones, phenol derivatives, as well as primary aromatic and heterocyclic amines, respectively. Most of the synthesized compounds were screened for their antifungal activity
-
Design and synthesis of pyrimidine-5-carbonitrile hybrids as COX-2 inhibitors: Anti-inflammatory activity, ulcerogenic liability, histopathological and docking studies作者:Abdallah M. Alfayomy、Salah A. Abdel-Aziz、Adel A. Marzouk、Montaser Sh. A. Shaykoon、Atsushi Narumi、Hiroyuki Konno、Sahar M. Abou-Seri、Fatma A.F. RagabDOI:10.1016/j.bioorg.2020.104555日期:2021.3Two new series of 1,3,4-oxadiazole and coumarin derivatives based on pyrimidine-5-carbonitrile scaffold have been synthesized and evaluated for their COX-1/COX-2 inhibitory activity. Compounds 10c, 10e, 10h-j, 14e-f, 14i and 16 were found to be the most potent and selective inhibitors of COX-2 (IC50 0.041-0.081 μM, SI 139.74-321.95). Eight compounds were further investigated for their in vivo anti-inflammatory合成了两个新系列的基于 pyrimidine-5-carbonitrile 支架的 1,3,4-恶二唑和香豆素衍生物,并评估了它们的 COX-1/COX-2 抑制活性。发现化合物10c、10e、10h-j、14e-f、14i和16是最有效和选择性的 COX-2 抑制剂(IC 50 0.041-0.081 μM,SI 139.74-321.95)。进一步研究了八种化合物的体内抗炎活性。最活跃的衍生物10c , 10j和14e显示出优于参考药物塞来昔布的体内抗炎活性(水肿抑制百分比 39.3-48.3,1 小时;58.4-60.5,2 小时;70.8-83.2,3 小时;78.9-89.5,4 小时)(水肿抑制百分比 38.0) ,1 小时;48.8、2 小时;58.4、3 小时;65.4、4 小时)。还测试了这些衍生物的致溃疡倾向,化合物10j与塞来昔布相比表现出更好的安全性,而10c和14e表现出轻度损伤。COX-2
表征谱图
-
氢谱1HNMR
-
质谱MS
-
碳谱13CNMR
-
红外IR
-
拉曼Raman
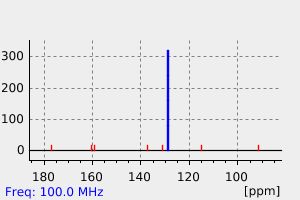
-
峰位数据
-
峰位匹配
-
表征信息
同类化合物
(S)-3-(2-(二氟甲基)吡啶-4-基)-7-氟-3-(3-(嘧啶-5-基)苯基)-3H-异吲哚-1-胺
(6-羟基嘧啶-4-基)乙酸
(4,5-二甲氧基-1,2,3,6-四氢哒嗪)
鲁匹替丁
马西替坦杂质7
马西替坦杂质4
马西替坦杂质
马西替坦原料药杂质D
马西替坦原料药杂质B
马西替坦
顺式-4-{[5-溴-2-(2,5-二甲基-1H-吡咯-1-基)-6-甲基嘧啶-4-基]氨基}环己醇
非沙比妥
非巴氨酯
非尼啶醇
青鲜素钾盐
雷特格韦钾盐
雷特格韦相关化合物E(USP)
雷特格韦杂质8
雷特格韦EP杂质H
雷特格韦-RT9
雷特格韦
阿西莫司杂质3
阿西莫司
阿脲四水合物
阿脲一水合物
阿维霉素
阿米美啶
阿米洛利
阿米妥钠
阿洛巴比妥
阿普瑞西他滨
阿普比妥
阿巴卡韦相关化合物B(USP)
阿卡明
阿伐那非杂质V
阿伐那非杂质1
阿伐那非杂质
阿伐那非中间体
阿伐那非
铂(2+)二氯化6-甲基-1,3-二{2-[(2-甲基丙基)硫烷基]乙基}嘧啶-2,4(1H,3H)-二酮(1:1)
钴1,2,3,6-四氢-2,6-二氧代嘧啶-4-羧酸酯(1:2)
钠5-烯丙基-4,6-二氧代-1,4,5,6-四氢-2-嘧啶醇酸酯
钠5-乙基-4,6-二氧代-1,4,5,6-四氢-2-嘧啶醇酸酯
钠5-(2-溴丙-2-烯基)-5-丁烷-2-基-4,6-二氧代-1H-嘧啶-2-醇
醌肟腙
酒石酸噻吩嘧啶
那可比妥
辛基2,6-二氧代-1,2,3,6-四氢-4-嘧啶羧酸酯
赛乐西帕杂质3
赛乐西帕KSM3

联系我们



关注我们

公众号

