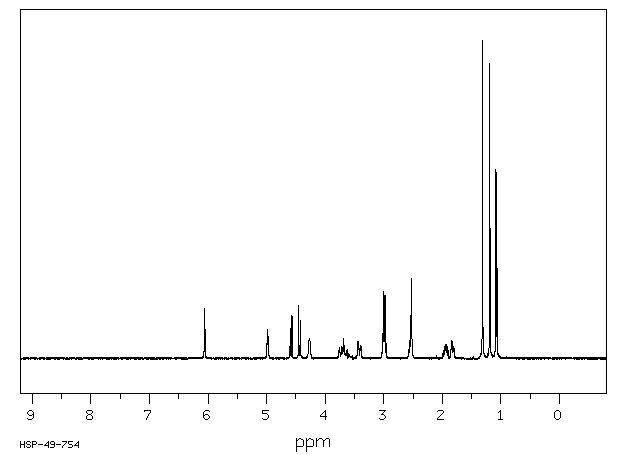
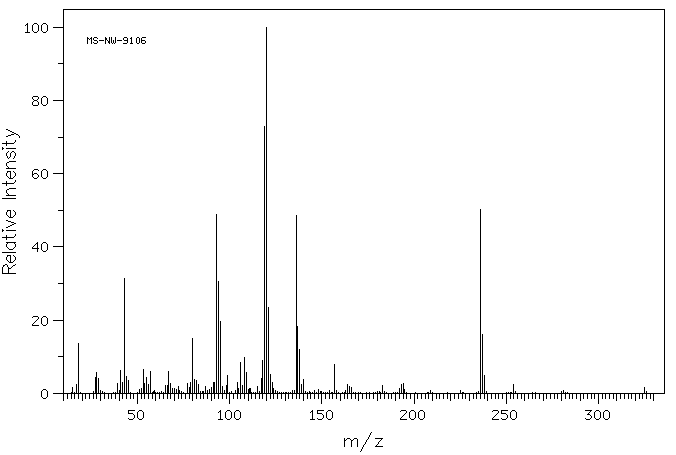
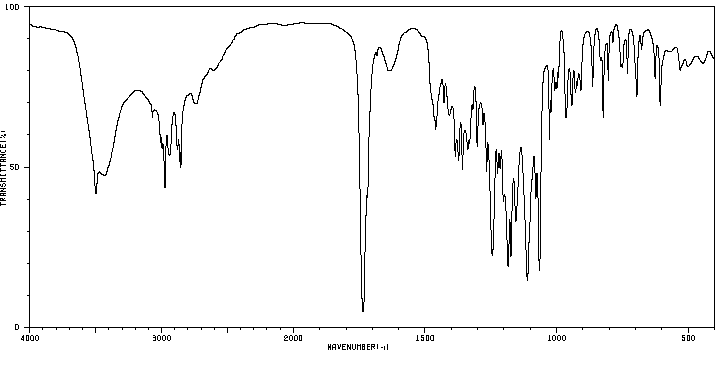
Studies with monocrotaline have confirmed the formation of pyrrolic metabolites by the mixed-function oxidase system of the microsomal fraction of rat liver. Dehydromonocrotaline (monocrotaline pyrrole) is highly cytotoxic, producing pulmonary, cardiac, vascular and hepatic lesions similar to those produced by the parent alkaloid. It is a highly reactive alkylating agent which, on formation within the cell, reacts immediately with cell constituents to give soluble or bound secondary metabolites or hydrolyzes to the dehydroaminoalcohol, dehydroretronecine.
Using microsomes from livers of phenobarbital-pretreated male rats, all 13 alkaloids tested were metabolized to n-oxide and pyrrole formation. The 2 pathways appeared to be independent. Ratio of n-oxide to pyrrolic metabolites varied, depending on type of ester: it was highest for open diester alkaloids and lowest for 12-membered macrocyclic diesters and for monoesters. Monocrotaline was one of those tested.
The comparative metabolism of the pyrrolizidine alkaloid, (14)C monocrotaline, was studied using rat and guinea pig hepatic microsomes. ... Esterase hydrolysis accounted for 92% of the metabolism in the guinea pig; the rat displayed no esterase activity. This result may explain the guinea pig's resistance to pyrrolizidine alkaloid toxicity. Dehydropyrrole was found to be the major pyrrolic metabolite in the guinea pig, although colorimetric analysis indicated multiple pyrrolic moieties in the rat microsomal incubations.
This report demonstrates that an Ehrlich reagent positive metabolite of monocrotaline and senecionine is excreted in the urine of male rats as an N-acetylcysteine conjugate of (+/-)-6,7-dihydro-7-hydroxy-1-hydroxymethyl-5H-pyrrolizine ... This finding suggests that reactive metabolites of pyrrolizidine alkaloids generated in the liver can survive the aqueous environment of the circulatory system as glutathione conjugates or mercapturic acids.
No data are available in humans. Sufficient evidence of carcinogenicity in animals. OVERALL EVALUATION: Group 2B: The agent is possibly carcinogenic to humans.
来源:International Agency for Research on Cancer (IARC)
毒理性
致癌物分类
国际癌症研究机构(IARC)致癌物分类:2B组:可能对人类致癌
IARC Carcinogenic Classes:Group 2B: Possibly carcinogenic to humans
来源:International Agency for Research on Cancer (IARC)
毒理性
致癌物分类
国际癌症研究机构专著:第10卷:(1976年)一些天然存在的物质
IARC Monographs:Volume 10: (1976) Some Naturally Occurring Substances
来源:International Agency for Research on Cancer (IARC)
毒理性
副作用
职业性肝毒素 - 第二性肝毒素:在职业环境中的毒性效应潜力是基于人类摄入或动物实验的中毒案例。
Occupational hepatotoxin - Secondary hepatotoxins: the potential for toxic effect in the occupational setting is based on cases of poisoning by human ingestion or animal experimentation.
来源:Haz-Map, Information on Hazardous Chemicals and Occupational Diseases
After sc administration of monocrotaline /in rats/, 50-70% of the dose was found in urine as unchanged monocrotaline... monocrotaline (or metabolite) concentration were highest in the liver, kidney and stomach.
The pyrrolizidine alkaloid monocrotaline has been shown to cause hepatic necrosis and pulmonary hypertension in the rat. To better understand the mechanism of action, tissue distribution and covalent binding studies were conducted at 4 and 24 hr following administration of (14)C monocrotaline (60 mg/kg, 200 microCi/kg, sc). For the 4 hr study, the levels of monocrotaline equivalents were 85, 74, 67, 36, and 8 nmol/g of tissue for RBC, liver, kidney, lung, and plasma, respectively, while the covalent binding levels were 125, 132, 39, 64, 44 pmol/mg of protein for tissues as listed above. The 24 hr tissue distribution levels were 49, 25, 9, 10, 2 nmol/g of tissue for RBC, liver, kidney, lung, and plasma, respectively, while covalent binding was 74, 28, and 55 pmol/mg of protein for liver, kidney, and lung, respectively. We also studied the kinetics of (14)C monocrotaline (60 mg/kg, 10 microCi/kg, iv), which demonstrated rapid elimination of radioactivity with approximately 90% recovery of the injected radioactivity in the urine and bile by 7 hr. The plasma levels of radioactivity dropped from 113 nmol/g of monocrotaline equivalents to 11 nmol/g at 7 hr while RBC levels decreased from 144 to only 81 nmol/g at the same time point. The apparent retention of monocrotaline equivalents in the RBC suggests that this organ may act as the carrier of metabolites from the liver to other organs including the lung and may play a role in the pulmonary toxicity.
The present invention relates to an N-substituted benzothiophenesulfonamide derivative or a pharmaceutically acceptable salt thereof and applications thereof. Furthermore, it provides an agent for preventing or treating cardiac or circulatory disease and so on caused by abnormal increase of production of angiotensin II or endothelin I based on chymase activity, or by activation of mast cell, and an agent for preventing adhesion after surgery, wherein the agent has a selective inhibitory action on chymase.
The present invention concerns a new process for the synthesis of aminaphtone, which makes use of non-toxic solvents and reagents, under mild reaction and temperature conditions. The aminaphtone obtained with the method of the present invention also has a purity of at least 98% in weight. The method comprises the following steps: a) epoxidating menadione 1 to provide epoxide 2, b) acidifying epoxide 2 to provide hydroxynaphthoquinone 3, c) esterifying between hydroxynaphthoquinone 3 and 4-aminobenzoyl chloride to obtain compound 4, and d) reducing compound 4 in the presence of a reducing agent in water to obtain aminaphtone.
A compound represented by the following formula (1) or a salt thereof:
wherein R
1
represents hydrogen atom, a halogen atom and the like; R
2
represents hydrogen atom, a halogen atom, a C
1-6
alkyl group and the like; and R
3
represents —O—X—C(A
1
)(A
11
)—C(A
2
)(A
2l
)—N(A
3l
)(A
3
)(X represents propylene group etc., A
11
and A
21
represent hydrogen atom, or a C
1-6
alkyl group, A
31
represents a C
1-6
alkyl group substituted with hydroxyl group, or hydrogen atom, and A
1
, A
2
, and A
3
represent hydrogen atom, a C
1-6
alkyl group and the like) and the like, which has an inhibitory activity on the phosphorylation of myosin regulatory light chain, and is useful for treatment of diseases relating to contraction of various cells and the like.
A novel compound represented by the following formula (1) or a salt thereof [wherein R
1
, R
5
, R
6
, R
7
, and R
8
represent hydrogen atom, a halogen atom, hydroxyl group, an alkyl group, an alkenyl group and the like; X
1
. . . X
2
represents —CH(R
2
)—CH(R
3
)—, —CH(R
2
)—CH(R
3
)—CH(R
4
)—, —C(R
2
)═C(R
3
)—, or —C(R
2
)═C(R
3
)—CH(R
4
)—(R
2
, R
3
, and R
4
represent hydrogen atom, or an alkyl group); A
1
, A
11
, A
2
, and A
21
represent hydrogen atom, or an alkyl group; Y represents —CH(A
3
)-, —CH(A
3
)-C(A
4
)(A
41
)-, —CH(A
3
)-C(A
4
)(A
41
)-C(A
5
)(A
51
)-, or a single bond (A
3
, A
4
, A
41
, A
5
, and A
51
represent hydrogen atom, or an alkyl group), and Z represents hydroxyl group, or —N(A
6
)(A
61
)(A
6
represents hydrogen atom, or an alkyl group, and A
61
represents hydrogen atom, an alkyl group, a substituted alkyl group and the like)], having an action of potently inhibiting phosphorylation of myosin regulatory light chain.
In one aspect, the invention relates to compounds having the formula XII:
where R
a
, R
b
, R
2
, R
7
, and X are as defined in the specification, or a pharmaceutically acceptable salt thereof. The compounds described herein are prodrugs of compounds having neprilysin inhibition activity. In another aspect, the invention relates to pharmaceutical compositions comprising these compounds; methods of using these compounds; and processes and intermediates for preparing these compounds.
在一方面,本发明涉及具有公式XII的化合物:
其中R
a
,R
b
,R
2
,R
7
和X如说明书中所定义,或其药用可接受的盐。所述的化合物是具有中性粒细胞弹性蛋白酶抑制活性的化合物的前药。在另一方面,本发明涉及包含这些化合物的药物组合物;使用这些化合物的方法;以及制备这些化合物的过程和中间体。