
4-(苄氧基)苯基异硫氰酸酯 | 139768-71-1
中文名称
4-(苄氧基)苯基异硫氰酸酯
中文别名
4-苄氧基苯基硫代异氰酸酯;4-苄氧基苯基异硫氰酸酯
英文名称
4-benzyloxyphenyl isothiocyanate
英文别名
1-isothiocyanato-4-phenylmethoxybenzene

CAS
139768-71-1
化学式
C14H11NOS
mdl
——
分子量
241.313
InChiKey
OQXRBXAFSXVCCO-UHFFFAOYSA-N
BEILSTEIN
——
EINECS
——
-
物化性质
-
计算性质
-
ADMET
-
安全信息
-
SDS
-
制备方法与用途
-
上下游信息
-
文献信息
-
表征谱图
-
同类化合物
-
相关功能分类
-
相关结构分类
物化性质
-
熔点:62 °C
-
沸点:392.9±25.0 °C(Predicted)
-
密度:1.09±0.1 g/cm3(Predicted)
-
稳定性/保质期:
如果按照规格正确使用和储存,则不会发生分解,目前没有已知的危险反应。
应避免与氧化物、水分等接触。
计算性质
-
辛醇/水分配系数(LogP):5.2
-
重原子数:17
-
可旋转键数:4
-
环数:2.0
-
sp3杂化的碳原子比例:0.07
-
拓扑面积:53.7
-
氢给体数:0
-
氢受体数:3
安全信息
-
危险等级:8
-
危险品标志:T
-
危险类别码:R20/22,R36/37/38
-
海关编码:2930909090
-
包装等级:III
-
危险类别:8
-
安全说明:S26,S36/37/39
-
危险品运输编号:UN 2923
-
储存条件:请将贮藏器保持密封,并存放在阴凉、干燥处。确保工作环境有良好的通风或排气设施。
SDS
上下游信息
-
上游原料
中文名称 英文名称 CAS号 化学式 分子量 4-苄氧基苯胺 p-benzyloxyaniline 6373-46-2 C13H13NO 199.252 4-苄氧基-1-硝基苯 benzyl 4-nitrophenyl ether 1145-76-2 C13H11NO3 229.235 -
下游产品
中文名称 英文名称 CAS号 化学式 分子量 1-(4-苄氧基苯基)-2-硫脲 N-(4-benzyloxyphenyl)thiourea 65069-53-6 C14H14N2OS 258.344
反应信息
-
作为反应物:参考文献:名称:Structure–activity relationship analysis of a novel necroptosis inhibitor, Necrostatin-5摘要:Necrostatin-5 (Nec-5) is a novel potent small-molecule inhibitor of necroptosis structurally distinct from previously described Necrostatin-1 (Nec-1), and therefore, represents a new direction for the inhibition of this cellular caspase-independent necrotic cell death mechanism. Here, we describe a series of structural modifications of Nec-5 and the structure-activity relationship (SAR) of Nec-5 series in inhibiting necroptosis.DOI:10.1016/j.bmcl.2006.11.056
-
作为产物:描述:参考文献:名称:作为新型人二氢乳清酸脱氢酶抑制剂的 4-噻唑烷酮衍生物的结构优化和构效关系摘要:人类二氢乳清酸脱氢酶(hDHODH)是开发免疫抑制药物的有吸引力的靶标之一,也是抗癌药物和抗白血病药物的潜在靶标。开发有前景的 hDHODH 抑制剂的需求量很大。基于我们之前报道的4-噻唑烷酮衍生物的独特结合模式,通过分子对接方法,设计并合成了三个新系列的4-噻唑烷酮衍生物作为hDHODH抑制剂。研究了初步的构效关系。联苯系列化合物9和酰胺系列化合物37的IC50值分别为1.32 μM和1.45 μM。该研究将为hDHODH抑制剂新结构的研究提供有价值的参考。DOI:10.3390/molecules24152780
文献信息
-
An automated, polymer-assisted strategy for the preparation of urea and thiourea derivatives of 15-membered azalides as potential antimalarial chemotherapeutics作者:Antun Hutinec、Renata Rupčić、Dinko Žiher、Kirsten S. Smith、Wilbur Milhous、William Ellis、Colin Ohrt、Zrinka Ivezić SchönfeldDOI:10.1016/j.bmc.2011.01.030日期:2011.3series of 15-membered azalide urea and thiourea derivatives has been synthesized and evaluated for their in vitro antimalarial activity against chloroquine-sensitive (D6), chloroquine/pyremethamine resistant (W2) and multidrug resistant (TM91C235) strains of Plasmodium falciparum. We have developed an effective automated synthetic strategy for the rapid synthesis of urea/thiourea libraries of a macrolide
-
Structure-Based Design, Parallel Synthesis, Structure−Activity Relationship, and Molecular Modeling Studies of Thiocarbamates, New Potent Non-Nucleoside HIV-1 Reverse Transcriptase Inhibitor Isosteres of Phenethylthiazolylthiourea Derivatives作者:Angelo Ranise、Andrea Spallarossa、Sara Cesarini、Francesco Bondavalli、Silvia Schenone、Olga Bruno、Giulia Menozzi、Paola Fossa、Luisa Mosti、Massimiliano La Colla、Giuseppina Sanna、Marta Murreddu、Gabriella Collu、Bernardetta Busonera、Maria Elena Marongiu、Alessandra Pani、Paolo La Colla、Roberta LoddoDOI:10.1021/jm049252r日期:2005.6.1In this paper we describe our structure-based ligand design, synthetic strategy, and structure-activity relationship (SAR) studies that led to the identification of thiocarbamates (TCs), a novel class of non-nucleoside reverse transcriptase inhibitors (NNRTIs), isosteres of phenethylthiazolylthiourea (PETT) derivatives. Assuming as a lead compound O-[2-(phthalimido)ethyl]phenylthiocarbamate 12, one在本文中,我们描述了我们基于结构的配体设计,合成策略和结构与活性关系(SAR)的研究,这些研究导致了硫代氨基甲酸酯(TC)的鉴定,这是一类新型的非核苷类逆转录酶抑制剂(NNRTIs),等位基因苯乙基噻唑基硫脲(PETT)衍生物的制备。假设O- [2-(邻苯二甲酰亚胺基)乙基]苯基硫代氨基甲酸酯为先导化合物,前述酰基硫代氨基甲酸酯的前体之一(Ranise,A .;等人,J。Med。Chem。2003,46,768-781) ,通过平行合成制备了两个目标溶液相TC库。领先的优化策略导致对位取代的TC 31、33、34、39、40、41、44、45和50在纳摩尔浓度的基于MT-4的测定中对野生型HIV-1具有活性( EC50范围:0.04-0.01 microM)。最有效的同类物50(EC50 = 0。01 microM)在邻苯二甲酰亚胺部分的第4位带有一个甲基,在N-苯环的对位带有一个硝基。大
-
Highly <i>Z</i>-selective synthesis of 1,3-oxathiol-2-ylidenes and 4-methylene-oxazolidine-2-thiones <i>via</i> atom-specific 5-<i>exo-dig</i> cyclization of propargyl alcohol with isothiocyanate作者:S. Antony Savarimuthu、D. G. Leo Prakash、S. Augustine Thomas、Thirumanavelan Gandhi、Mrinal K. BeraDOI:10.1039/d0ob00083c日期:——internal propargyl alcohol to produce (Z)-1,3-oxathiol-2-ylidenes and (Z)-N-(Z)-4-ethylidene-1,3-oxathiolan-2-ylidenes from secondary and primary propargyl alcohols, respectively. The formation of high Z-selectivity in the imine motif and alkene is the highlight of this new method as multiple selectivities over C[double bond, length as m-dash]N and C[double bond, length as m-dash]C in a single system are syntheticallyDBU介导的异硫氰酸酯和炔丙醇的5-exo-dig环化反应可形成有价值的杂环化合物。发现异硫氰酸酯的亲核性的不同模式(S-选择性或N-选择性)取决于炔丙醇的取代模式。对末端炔丙醇和异硫氰酸酯进行N-亲核攻击,得到3-取代的4-亚甲基恶唑烷-2-硫酮。相反,观察到内部炔丙醇的独家S亲核环化反应产生(Z)-1,3-氧杂硫醇-2-亚烷基和(Z)-N-(Z)-4-亚乙基-1,3-氧杂硫杂环戊烷-分别来自仲和伯炔丙醇的2-亚烷基。在亚胺基序和烯烃中高Z选择性的形成是这种新方法的亮点,因为在C [双键,在单个系统中,如m-N和C双键的长度,如m-C的长度在合成上是非常具有挑战性的。亚胺和烯烃中的Z-选择性可以分别归因于电子和空间因素。
-
Biphenyl-substituted guanidine derivatives useful as hypoglycaemic agents申请人:The Boots Company公开号:US05302720A1公开(公告)日:1994-04-12Compounds of formula I ##STR1## and their salts in which R.sub.1 is optionally substituted phenyl, R.sub.2 is alkyl, cycloalkyl or optionally substituted amino, or R.sub.2 and R.sub.3 together with the nitrogen and carbon atoms to which they are attached form an optionally substituted heterocyclic ring or R.sub.3 and R.sub.4 together with the nitrogen atom to which they are attached form an optionally substituted heterocyclic ring and R.sub.5 is H, halo, alkyl, alkoxy, trifluoromethyl or a group of formula S(O).sub.m R.sub.8 in which m is 0, 1 or 2 and R.sub.8 is alkyl have utility in the treatment of diabetes particularly in the treatment of hyperglycaemia.
-
Synthesis and evaluation of new thiourea derivatives as antitumor and antiangiogenic agents作者:Wenjing Bai、Jianxin Ji、Qiang Huang、Wei WeiDOI:10.1016/j.tetlet.2020.152366日期:2020.10A series of novel thiourea derivatives were synthesized and evaluated by biological activities. Among them, compound 10e containing 3,5-bis(trifluoromethyl)phenyl moiety (R1) at the terminal thiourea and phenylamino (R2) at the terminal acyl position showed the best cytotoxic activities against seven cancer cell lines (NCI-H460, Colo-205, HCT116, MDA-MB-231, MCF-7, HepG2, PLC/PRF/5) and HUVECs. Moreover
表征谱图
-
氢谱1HNMR
-
质谱MS
-
碳谱13CNMR
-
红外IR
-
拉曼Raman
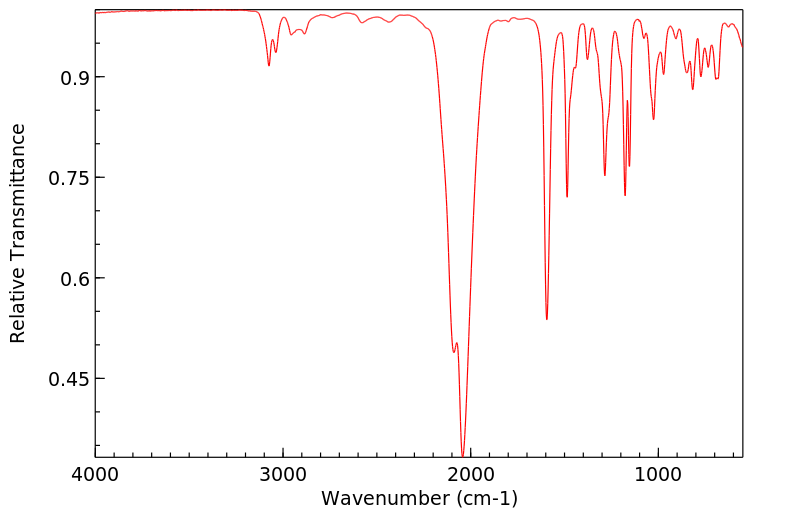
-
峰位数据
-
峰位匹配
-
表征信息
同类化合物
(R)-3-(叔丁基)-4-(2,6-二异丙氧基苯基)-2,3-二氢苯并[d][1,3]氧杂磷杂环戊烯
(2S,3R)-3-(叔丁基)-2-(二叔丁基膦基)-4-甲氧基-2,3-二氢苯并[d][1,3]氧杂磷杂戊环
(2S,2''S,3S,3''S)-3,3''-二叔丁基-4,4''-二甲氧基-2,2'',3,3''-四氢-2,2''-联苯并[d][1,3]氧杂磷杂戊环
(2R,2''R,3R,3''R)-3,3''-二叔丁基-4,4''-二甲氧基-2,2'',3,3''-四氢-2,2''-联苯并[d][1,3]氧杂磷杂戊环
(2-氟-3-异丙氧基苯基)三氟硼酸钾
(+)-6,6'-{[(1R,3R)-1,3-二甲基-1,3基]双(氧)}双[4,8-双(叔丁基)-2,10-二甲氧基-丙二醇
麦角甾烷-6-酮,2,3,22,23-四羟基-,(2a,3a,5a,22S,23S)-
鲁前列醇
顺式6-(对甲氧基苯基)-5-己烯酸
顺式-铂戊脒碘化物
顺式-四氢-2-苯氧基-N,N,N-三甲基-2H-吡喃-3-铵碘化物
顺式-4-甲氧基苯基1-丙烯基醚
顺式-2,4,5-三甲氧基-1-丙烯基苯
顺式-1,3-二甲基-4-苯基-2-氮杂环丁酮
非那西丁杂质7
非那西丁杂质3
非那西丁杂质22
非那西丁杂质18
非那卡因
非布司他杂质37
非布司他杂质30
非布丙醇
雷诺嗪
阿达洛尔
阿达洛尔
阿莫噁酮
阿莫兰特
阿维西利
阿索卡诺
阿米维林
阿立酮
阿曲汀中间体3
阿普洛尔
阿普斯特杂质67
阿普斯特中间体
阿普斯特中间体
阿托西汀EP杂质A
阿托莫西汀杂质24
阿托莫西汀杂质10
阿托莫西汀EP杂质C
阿尼扎芬
阿利克仑中间体3
间苯胺氢氟乙酰氯
间苯二酚二缩水甘油醚
间苯二酚二异丙醇醚
间苯二酚二(2-羟乙基)醚
间苄氧基苯乙醇
间甲苯氧基乙酸肼
间甲苯氧基乙腈
间甲苯异氰酸酯

联系我们



关注我们

公众号
