Two guinea pig models were used to study the anticonvulsant potency of diazepam, midazolam, and scopolamine against seizures induced by the nerve agents tabun, sarin, soman, cyclosarin, O-ethyl S-(2-(diisopropylamino)ethyl)methylphosphonothioate (VX), and O-isobutyl S-(2-diethylamino)ethyl)-methyl phosphonothioate (VR). Animals instrumented for electroencephalogram recording were pretreated with pyridostigmine bromide (0.026 mg/kg im) 30 min before challenge with 2 x LD50 (s.c.) of a nerve agent. In model A, atropine sulfate (2.0 mg/kg im) and pyridine-2-aldoxime methylchloride (2-PAM; 25.0 mg/kg im) were given 1 min after nerve agent challenge, and the tested anticonvulsant was given (im) 5 min after seizure onset. In model B, a lower dose of atropine sulfate (0.1 mg/kg im) was given along with 2-PAM 1 min after nerve agent challenge, and the anticonvulsant was given at seizure onset. With the lower dose of atropine, seizure occurrence increased to virtually 100% for all agents; the time to seizure onset decreased for sarin, cyclosarin, and VX; the signs of nerve agent intoxication were more severe; and coma resulted frequently with cyclosarin. The anticonvulsant ED50 doses for scopolamine or diazepam were, in general, not different between the two models, whereas the anticonvulsant ED50 values of midazolam increased 3- to 17-fold with the lower atropine dose. Seizure termination times were not systematically affected by the different doses of atropine. The order of anticonvulsant effectiveness within each model was scopolamine > or = midazolam > diazepam. The findings indicate that the dose of atropine given as antidotal therapy can significantly influence measures of nerve agent toxicity and responsiveness to anticonvulsant therapy.
Currently fielded treatments for nerve agent intoxication promote survival, but do not afford complete protection against either nerve agent-induced motor and cognitive deficits or neuronal pathology. The use of human plasma-derived butyrylcholinesterase (HuBuChE) to neutralize the toxic effects of nerve agents in vivo has been shown to both aid survival and protect against decreased cognitive function after nerve agent exposure. Recently, a commercially produced recombinant form of human butyrylcholinesterase (r-HuBuChE ...) expressed in the milk of transgenic goats has become available. This material is biochemically similar to plasma-derived HuBuChE in in vitro assays. The pharmacokinetic characteristics of a polyethylene glycol coated (pegylated) form of r-HuBuChE were determined in guinea pigs; the enzyme was rapidly bioavailable with a half-life (t1/2) and pharmacokinetic profile that resembled that of plasma-derived huBuChE. Guinea pigs were injected with 140 mg/kg (im) of pegylated r-HuBuChE 18 hr prior to exposure (sc) to 5.5xLD50 VX or soman. VX and soman were administered in a series of three injections of 1.5xLD50, 2.0xLD50, and 2.0xLD50, respectively, with injections separated by 2 hr. Pretreatment with pegylated r-HuBuChE provided 100% survival against multiple lethal doses of VX and soman. Guinea pigs displayed no signs of nerve agent toxicity following exposure. Assessments of motor activity, coordination, and acquisition of spatial memory were performed for 2 weeks following nerve agent exposure. There were no measurable decreases in motor or cognitive function during this period. In contrast, animals receiving 1.5xLD50 challenges of soman or VX and treated with standard atropine, 2-PAM, and diazepam therapy showed 50 and 100% survival, respectively, but exhibited marked decrements in motor function and, in the case of GD, impaired spatial memory acquisition. The advances in this field have resulted in the decision to select both the plasma-derived and the recombinant form of BuChE for advanced development and transition to clinical trials. ...
Organophosphorus compounds such as nerve agents inhibit, practically irreversibly, cholinesterases by their phosphorylation in the active site of these enzymes. Current antidotal treatment used in the case of acute nerve agent intoxications consists of combined administration of anticholinergic drug (usually atropine) and acetylcholinesterase (AChE, EC 3.1.1.7) reactivator (HI-6, obidoxime, pralidoxime), which from a chemical view is a derivative from the group of pyridinium or bispyridinium aldoximes (commonly called "oxime"). Oximes counteract acetylcholine increase, resulting from AChE inhibition. In the human body environment these compounds are powerful nucleophiles and are able to break down the bond between AChE and nerve agent molecule. This process leads to renewal of enzyme functionality -- to its reactivation. The usefulness of oxime in the reactivation process depends on its chemical structure and on the nerve agent whereby AChE is inhibited. Due to this fact, selection of suitable reactivator in the treatment of intoxications is very important. This work compared differences in the in vitro inhibition potency of VX and Russian VX on rat, pig and human brain, and subsequently we have tested reactivation of rat brain cholinesterase inhibited by these agents using oxime HI-6, obidoxime, pralidoxime, trimedoxime and methoxime. The results showed that no major differences in the reactivation process of both VX and Russian VX-inhibited cholinesterase. The similarity in reactivation was caused by analogous chemical structure of either nerve agent; and that oxime HI-6 seems to be the most effective reactivator tested, which confirms that HI-6 is currently the most potent reactivator of AChE inhibited by nerve agents...
... The present work was designed to assess the ability of butyrylcholinesterase, purified from human serum (HuBChE), to prevent the toxicity induced by soman and VX in rhesus monkeys. The consistency of the data across species was then evaluated as the basis for the extrapolation of the data to humans. The average mean residence time of the enzyme in the circulation of monkeys following an intravenous loading was 34 hr. High bioavailability of HuBChE in blood (>80%) was demonstrated after intramuscular injection. A molar ratio of HuBChE:OP approximately 1.2 protected against an i.v. bolus injection of 2.1 x LD50 VX, while a ratio of 0.62 was sufficient to protect monkeys against an i.v. dose of 3.3 x LD50 of soman, with no additional postexposure therapy. A remarkable protection was also seen against soman-induced behavioral deficits detected in the performance of a spatial discrimination task. The consistency of the results across several species offers a reliable prediction of both the stoichiometry of the scavenging and the extent of prophylaxis with HuBChE against nerve agent toxicity in humans.
The purpose of this study was to evaluate the efficacy of a novel barrier cream formulation at reducing the percutaneous toxicity of a 2xLD(50) liquid challenge of nerve agent (VX) ... in vitro and in vivo using the domestic pig. Pretreatment of the (inner ear skin) exposure site with barrier cream eliminated mortality, reduced cholinesterase inhibition and prevented any physiological or biochemical signs of intoxication. In contrast, untreated animals exposed to VX exhibited severe signs of intoxication, near total AChE inhibition and generally died within the (3 hr) exposure period (5/6 animals). Application of the barrier cream caused a significant decrease in the area of skin contaminated by VX. It was tentatively concluded that spreading was predominantly a surface phenomena (possibly mediated by capillary movement of the agent through the microrelief or between hair follicles) with little or no contribution from lateral diffusion within the stratum corneum. There was a disparity between the in vitro and in vivo skin absorption measurements that was ascribed to the absence of systemic clearance in vitro. However, both models indicated a substantial reservoir of VX within the skin ...
... In order to obtain basic information on the toxicokinetics of (+/-)-VX, i.e., under conditions of 100% bioavailability, the blood levels of this agent were measured in hairless guinea pigs at iv doses corresponding with 1 and 2 x LD50. The derived AUCs indicate a reasonable linearity of the toxicokinetics with dose. Also, the toxicokinetics in marmoset primates was studied at an absolute iv dose corresponding with 1 x LD50 in the hairless guinea pig which led to approximately the same levels of (+/-)-VX in blood as observed at 2 x LD50 in the hairless guinea pig. Finally, the toxicokinetics of (+/-)-VX were measured in hairless guinea pigs via the ... percutaneous route at a dose corresponding with 1 x LD50 (pc). Large variations were observed between individual animals in the rate of penetration of (+/-)-VX and in concomitant progression of AChE inhibition in blood of these animals. Blood levels of (+/-)-VX increased gradually over a 6-hr period of time. After a 7-hr penetration period, the total AUC corresponded with 2.5% bioavailability relative to iv administration. In contrast with the G-agents C(+/-)P(+/-)-soman and (+/-)-sarin, stereospecificity in the sequestration of the two enantiomers of (+/-)-VX is not a prominent phenomenon. It appears that (+/-)-VX is substantially more persistent in vivo than the two G-agents. This persistence may undermine the efficacy of pretreatment with carbamates of percutaneous intoxication in particular due to gradual replacement of carbamate on AChE by (+/-)-VX, whereas classical treatment of intoxication with oximes is hampered by the short persistence of oximes relative to the agent.
... The purpose of the present study was to measure and compare the skin absorption kinetics of VX in vitro using pig, human and guinea pig skin to highlight any potential species differences in skin permeability. When undiluted VX was applied directly to the skin, the permeability of guinea pig skin was approximately 7-fold greater than human skin. There was no significant difference in the permeability of pig and human skin. When VX diluted with isopropyl alcohol was applied to the skin, the permeability of guinea pig skin was approximately 4-fold greater than human skin. There was no significant difference in the permeability of pig and human skin. From this data it may be inferred that dermatomed, abdominal pig skin is an appropriate model for the human skin absorption of VX.
Studies of the penetration of liquid VX through skin were conducted with Service volunteers in November 1960 ... . Drops of radioactively-tagged VX were placed on the skin in one of three locations: the back, the forearm, or the palm of the hand. The chosen area, excepting the palm, was shaved with an electric razor and a ring was attached to the skin. The rings used on the forearm and the palm were made of polythene with an internal diameter of 2.5 cm; those on the back were made of brass measuring 7 cm in diameter. VX was placed on the skin inside the ring in drops of 2 - 4 ug. Nylon film was stuck over the top of the ring. The assembly and VX were left in place for 6-10 hours, during which time a Geiger counter was used to take measurements. The assembly was then removed and the skin decontaminated, first by stripping with adhesive tape and then by washing with isopropanol and soapy water. ... According to the volunteer records, 16 men took part in this study. ... ChE depressions observed were small, varying from 1.9% to 3.9%. After a brief delay the penetration of VX through the forearm and the back was "regular and rapid": on average, 8% of the VX dose penetrated through back skin and 15% through forearm skin in 8 hours.
Synthesis of macroscopic monolithic metal–organic gels for ultra-fast destruction of chemical warfare agents
作者:Chuan Zhou、Shouxin Zhang、Hongjie Pan、Guang Yang、Lingyun Wang、Cheng-an Tao、Heguo Li
DOI:10.1039/d1ra01703a
日期:——
for the destruction of CWAs. We found that the UiO-66-NH2 xerogel with a larger pore size and a higher surface area than the UiO-66-NH2 powder possessed better degradability of 2-chloroethyl ethyl sulfide (2-CEES), which is a sulfur mustard simulant. These UiO-66-X xerogels exhibit outstanding performance for decomposing CWAs. The half-lives of vesicant agent sulfur mustard (HD) and nerve agent O-ethyl
Facile Hydrolysis-Based Chemical Destruction of the Warfare Agents VX, GB, and HD by Alumina-Supported Fluoride Reagents
作者:E. Gershonov、I. Columbus、Y. Zafrani
DOI:10.1021/jo8019972
日期:2009.1.2
A facile solvent-free hydrolysis (chemicaldestruction) of the warfareagentsVX (O-ethyl S-2-(diisopropylamino)ethyl methylphosphonothioate), GB (O-isopropyl methylphosphonofluoridate or sarin), and HD (2,2′-dichloroethyl sulfide or sulfur mustard) upon reaction with various solid-supported fluoride reagents is described. These solid reagents include different alumina-based powders such as KF/Al2O3
简便的无溶剂水解剂(战争破坏剂VX(O-乙基S -2-(二异丙氨基氨基)乙基甲基硫代磷酸酯),GB(O-异丙基甲基氟代磷酸酯或沙林)和HD(2,2'-二氯乙基硫醚)描述了在与各种固体负载的氟化物试剂反应后产生的硫芥末或硫芥末)。这些固体试剂包括不同的氧化铝基粉末,例如KF / Al 2 O 3,AgF / KF / Al 2 O 3和KF / Al 2 O 3,它们富含所谓的配位不饱和氟化物离子(我们称其为ECUF- KF / Al 2 O 3)。当吸附在这些吸附剂上时,神经毒剂VX迅速水解(t 1/2范围在0.1-6.3 h之间)水解成相应的无毒膦酸主要成分EMPA(> 90%)和相对毒性的脱乙基VX(< 10%)。后者的副产物进一步水解为无毒的MPA产物(t 1/2范围为2.2-161 h)。发现反应速率和产物分布强烈地取决于KF / Al 2 O 3基质中氟离子的性质及其含水量。所研
Group 13 chelates in nerve gas agent and pesticide dealkylation
作者:Amitabha Mitra、David A. Atwood、Jeffrey Struss、Daniel J. Williams、Bradley J. McKinney、William R. Creasy、David J. McGarvey、H. Dupont Durst、Roderick Fry
DOI:10.1039/b717041f
日期:——
Schiff base boron and aluminium bromides have been used to cleave organophosphate nerve agents and pesticides and their simulants: salben(tBu)[BBr2]2 was very effective in cleaving the VX simulants EMPPT and DEPPT and nerve agent VX; salen(tBu)AlBr was effective in cleaving the nerve agents VX and Soman and the pesticideDiazinon.
Reaction of Nerve Agents with Phosphate Buffer at pH 7
作者:William R. Creasy、Roderick A. Fry、David J. McGarvey
DOI:10.1021/jp3024809
日期:2012.7.12
31P NMR. Phosphate causes faster reaction to the corresponding alkyl methylphosphonic acids, and produces a mixed phosphate/phosphonate compound as an intermediate reaction product. GB has the fastest reaction rate, with a bimolecular rate constant of 4.6 × 10–3 M–1s–1[PO43-]. The molar product branching ratio of GB acid to the pyro product (isopropyl methylphosphonate phosphate anhydride) is 1:1.4, independent
化学武器神经毒剂,包括异丙基甲基膦酰氟(GB或Sarin),频哪醇甲基膦酰氟(GD或Soman)和S-(2-二异丙基氨基乙基)O-乙基甲基膦酰硫(VX),在中等pH值的水溶液中反应缓慢。相对于蒸馏水或醋酸盐缓冲液,pH值为7的磷酸盐缓冲液中神经药的反应性增加。使用31 P NMR研究反应。磷酸盐导致与相应的烷基甲基膦酸的反应更快,并产生混合的磷酸盐/膦酸酯化合物作为中间反应产物。GB具有最快的反应速率,双分子速率常数为4.6×10 –3 M –1 s –1 [PO4 3- ]。GB酸与焦油产物(异丙基甲基膦酸酯磷酸酐)的摩尔产物支化比为1:1.4,与磷酸盐浓度无关,焦油产物继续反应得很慢,形成GB酸。焦烧产物在31 P NMR光谱中有两个双峰。GD的反应速率比GB慢,速率常数为1.26×10 –3 M –1 s –1 [PO 4 3- ]。VX的速率要慢得多,速率常数为1.39×10 –5
Oxidative detoxification of phosphonothiolates
作者:Yu Chu Yang、Linda L. Szafraniec、William T. Beaudry、Dennis K. Rohrbaugh
DOI:10.1021/ja00174a025
日期:1990.8
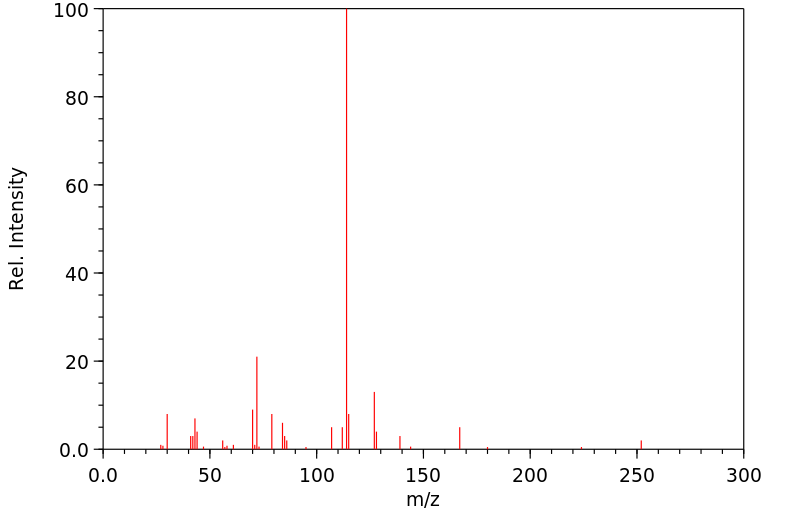
The chemical nerve agent O-ethyl S-[2-(diisopropylamino) ethyl] methylphosphonothiolate (VX) is an unusually selective oxidation substrate. Relative to the thiolo sulfur, the amino nitrogen was a more reactive oxidation site. The oxidation of VX and a phosphonothiolate derivative by a broad range of peroxygen compounds was examined in organic, polar organic, and aqueous solvents

![O-乙基-S-[2-(二异丙氨基)乙基]甲基硫代磷酸酯化学式](data:image/svg+xml;base64,PD94bWwgdmVyc2lvbj0iMS4wIiBlbmNvZGluZz0iVVRGLTgiPz4KPHN2ZyB4bWxucz0iaHR0cDovL3d3dy53My5vcmcvMjAwMC9zdmciIHhtbG5zOnhsaW5rPSJodHRwOi8vd3d3LnczLm9yZy8xOTk5L3hsaW5rIiB3aWR0aD0iMzAwIiBoZWlnaHQ9IjMwMCIgdmlld0JveD0iMCAwIDMwMCAzMDAiPgo8ZGVmcz4KPGc+CjxnIGlkPSJnbHlwaC0wLTAiPgo8cGF0aCBkPSJNIDAuNjcxODc1IDIuMzc1IEwgMC42NzE4NzUgLTkuNDY4NzUgTCA3LjM3NSAtOS40Njg3NSBMIDcuMzc1IDIuMzc1IFogTSAxLjQyMTg3NSAxLjYyNSBMIDYuNjI1IDEuNjI1IEwgNi42MjUgLTguNzAzMTI1IEwgMS40MjE4NzUgLTguNzAzMTI1IFogTSAxLjQyMTg3NSAxLjYyNSAiLz4KPC9nPgo8ZyBpZD0iZ2x5cGgtMC0xIj4KPHBhdGggZD0iTSAyLjY0MDYyNSAtOC43MDMxMjUgTCAyLjY0MDYyNSAtNS4wMTU2MjUgTCA0LjMxMjUgLTUuMDE1NjI1IEMgNC45MjU3ODEgLTUuMDE1NjI1IDUuMzk4NDM4IC01LjE3NTc4MSA1LjczNDM3NSAtNS41IEMgNi4wNjY0MDYgLTUuODIwMzEyIDYuMjM0Mzc1IC02LjI3MzQzOCA2LjIzNDM3NSAtNi44NTkzNzUgQyA2LjIzNDM3NSAtNy40NDE0MDYgNi4wNjY0MDYgLTcuODk0NTMxIDUuNzM0Mzc1IC04LjIxODc1IEMgNS4zOTg0MzggLTguNTM5MDYyIDQuOTI1NzgxIC04LjcwMzEyNSA0LjMxMjUgLTguNzAzMTI1IFogTSAxLjMxMjUgLTkuNzgxMjUgTCA0LjMxMjUgLTkuNzgxMjUgQyA1LjQwNjI1IC05Ljc4MTI1IDYuMjM0Mzc1IC05LjUzMTI1IDYuNzk2ODc1IC05LjAzMTI1IEMgNy4zNTkzNzUgLTguNTM5MDYyIDcuNjQwNjI1IC03LjgxNjQwNiA3LjY0MDYyNSAtNi44NTkzNzUgQyA3LjY0MDYyNSAtNS44OTA2MjUgNy4zNTkzNzUgLTUuMTYwMTU2IDYuNzk2ODc1IC00LjY3MTg3NSBDIDYuMjM0Mzc1IC00LjE3OTY4OCA1LjQwNjI1IC0zLjkzNzUgNC4zMTI1IC0zLjkzNzUgTCAyLjY0MDYyNSAtMy45Mzc1IEwgMi42NDA2MjUgMCBMIDEuMzEyNSAwIFogTSAxLjMxMjUgLTkuNzgxMjUgIi8+CjwvZz4KPGcgaWQ9ImdseXBoLTAtMiI+CjxwYXRoIGQ9Ik0gNy4xODc1IC05LjQ2ODc1IEwgNy4xODc1IC04LjE3MTg3NSBDIDYuNjc1NzgxIC04LjQxMDE1NiA2LjE5NTMxMiAtOC41ODU5MzggNS43NSAtOC43MDMxMjUgQyA1LjMxMjUgLTguODI4MTI1IDQuODgyODEyIC04Ljg5MDYyNSA0LjQ2ODc1IC04Ljg5MDYyNSBDIDMuNzUgLTguODkwNjI1IDMuMTkxNDA2IC04Ljc1IDIuNzk2ODc1IC04LjQ2ODc1IEMgMi40MTAxNTYgLTguMTg3NSAyLjIxODc1IC03Ljc4OTA2MiAyLjIxODc1IC03LjI4MTI1IEMgMi4yMTg3NSAtNi44NDM3NSAyLjM0NzY1NiAtNi41MTU2MjUgMi42MDkzNzUgLTYuMjk2ODc1IEMgMi44NjcxODggLTYuMDc4MTI1IDMuMzU5Mzc1IC01Ljg5ODQzOCA0LjA3ODEyNSAtNS43NjU2MjUgTCA0Ljg3NSAtNS41OTM3NSBDIDUuODYzMjgxIC01LjQwNjI1IDYuNTkzNzUgLTUuMDcwMzEyIDcuMDYyNSAtNC41OTM3NSBDIDcuNTMxMjUgLTQuMTI1IDcuNzY1NjI1IC0zLjQ5MjE4OCA3Ljc2NTYyNSAtMi43MDMxMjUgQyA3Ljc2NTYyNSAtMS43NTM5MDYgNy40NDUzMTIgLTEuMDM1MTU2IDYuODEyNSAtMC41NDY4NzUgQyA2LjE3NTc4MSAtMC4wNTQ2ODc1IDUuMjUgMC4xODc1IDQuMDMxMjUgMC4xODc1IEMgMy41NjI1IDAuMTg3NSAzLjA2NjQwNiAwLjEzMjgxMiAyLjU0Njg3NSAwLjAzMTI1IEMgMi4wMjM0MzggLTAuMDcwMzEyNSAxLjQ4NDM3NSAtMC4yMjY1NjIgMC45MjE4NzUgLTAuNDM3NSBMIDAuOTIxODc1IC0xLjc5Njg3NSBDIDEuNDYwOTM4IC0xLjQ5MjE4OCAxLjk4ODI4MSAtMS4yNjU2MjUgMi41IC0xLjEwOTM3NSBDIDMuMDE5NTMxIC0wLjk2MDkzOCAzLjUzMTI1IC0wLjg5MDYyNSA0LjAzMTI1IC0wLjg5MDYyNSBDIDQuNzgxMjUgLTAuODkwNjI1IDUuMzU5Mzc1IC0xLjAzNTE1NiA1Ljc2NTYyNSAtMS4zMjgxMjUgQyA2LjE3OTY4OCAtMS42Mjg5MDYgNi4zOTA2MjUgLTIuMDU0Njg4IDYuMzkwNjI1IC0yLjYwOTM3NSBDIDYuMzkwNjI1IC0zLjA4NTkzOCA2LjIzODI4MSAtMy40NjA5MzggNS45Mzc1IC0zLjczNDM3NSBDIDUuNjQ0NTMxIC00LjAwMzkwNiA1LjE2NDA2MiAtNC4yMDcwMzEgNC41IC00LjM0Mzc1IEwgMy42ODc1IC00LjUgQyAyLjY5NTMxMiAtNC42OTUzMTIgMS45ODQzNzUgLTUuMDAzOTA2IDEuNTQ2ODc1IC01LjQyMTg3NSBDIDEuMTA5Mzc1IC01LjgzNTkzOCAwLjg5MDYyNSAtNi40MjE4NzUgMC44OTA2MjUgLTcuMTcxODc1IEMgMC44OTA2MjUgLTguMDM1MTU2IDEuMTkxNDA2IC04LjcxODc1IDEuNzk2ODc1IC05LjIxODc1IEMgMi40MTAxNTYgLTkuNzE4NzUgMy4yNSAtOS45Njg3NSA0LjMxMjUgLTkuOTY4NzUgQyA0Ljc2OTUzMSAtOS45Njg3NSA1LjIzODI4MSAtOS45MjU3ODEgNS43MTg3NSAtOS44NDM3NSBDIDYuMTk1MzEyIC05Ljc1NzgxMiA2LjY4NzUgLTkuNjMyODEyIDcuMTg3NSAtOS40Njg3NSBaIE0gNy4xODc1IC05LjQ2ODc1ICIvPgo8L2c+CjxnIGlkPSJnbHlwaC0wLTMiPgo8cGF0aCBkPSJNIDUuMjgxMjUgLTguODkwNjI1IEMgNC4zMjAzMTIgLTguODkwNjI1IDMuNTYyNSAtOC41MzEyNSAzIC03LjgxMjUgQyAyLjQzNzUgLTcuMDkzNzUgMi4xNTYyNSAtNi4xMTMyODEgMi4xNTYyNSAtNC44NzUgQyAyLjE1NjI1IC0zLjY0NDUzMSAyLjQzNzUgLTIuNjcxODc1IDMgLTEuOTUzMTI1IEMgMy41NjI1IC0xLjI0MjE4OCA0LjMyMDMxMiAtMC44OTA2MjUgNS4yODEyNSAtMC44OTA2MjUgQyA2LjI1IC0wLjg5MDYyNSA3LjAxNTYyNSAtMS4yNDIxODggNy41NzgxMjUgLTEuOTUzMTI1IEMgOC4xNDA2MjUgLTIuNjcxODc1IDguNDIxODc1IC0zLjY0NDUzMSA4LjQyMTg3NSAtNC44NzUgQyA4LjQyMTg3NSAtNi4xMTMyODEgOC4xNDA2MjUgLTcuMDkzNzUgNy41NzgxMjUgLTcuODEyNSBDIDcuMDE1NjI1IC04LjUzMTI1IDYuMjUgLTguODkwNjI1IDUuMjgxMjUgLTguODkwNjI1IFogTSA1LjI4MTI1IC05Ljk2ODc1IEMgNi42NTYyNSAtOS45Njg3NSA3Ljc1MzkwNiAtOS41MDM5MDYgOC41NzgxMjUgLTguNTc4MTI1IEMgOS4zOTg0MzggLTcuNjYwMTU2IDkuODEyNSAtNi40MjU3ODEgOS44MTI1IC00Ljg3NSBDIDkuODEyNSAtMy4zMzIwMzEgOS4zOTg0MzggLTIuMTAxNTYyIDguNTc4MTI1IC0xLjE4NzUgQyA3Ljc1MzkwNiAtMC4yNjk1MzEgNi42NTYyNSAwLjE4NzUgNS4yODEyNSAwLjE4NzUgQyAzLjkwNjI1IDAuMTg3NSAyLjgwNDY4OCAtMC4yNjk1MzEgMS45ODQzNzUgLTEuMTg3NSBDIDEuMTYwMTU2IC0yLjEwMTU2MiAwLjc1IC0zLjMzMjAzMSAwLjc1IC00Ljg3NSBDIDAuNzUgLTYuNDI1NzgxIDEuMTYwMTU2IC03LjY2MDE1NiAxLjk4NDM3NSAtOC41NzgxMjUgQyAyLjgwNDY4OCAtOS41MDM5MDYgMy45MDYyNSAtOS45Njg3NSA1LjI4MTI1IC05Ljk2ODc1IFogTSA1LjI4MTI1IC05Ljk2ODc1ICIvPgo8L2c+CjxnIGlkPSJnbHlwaC0wLTQiPgo8cGF0aCBkPSJNIDguNjQwNjI1IC05LjAzMTI1IEwgOC42NDA2MjUgLTcuNjQwNjI1IEMgOC4xOTE0MDYgLTguMDQ2ODc1IDcuNzE4NzUgLTguMzUxNTYyIDcuMjE4NzUgLTguNTYyNSBDIDYuNzE4NzUgLTguNzY5NTMxIDYuMTc5Njg4IC04Ljg3NSA1LjYwOTM3NSAtOC44NzUgQyA0LjQ5MjE4OCAtOC44NzUgMy42NDA2MjUgLTguNTMxMjUgMy4wNDY4NzUgLTcuODQzNzUgQyAyLjQ1MzEyNSAtNy4xNjQwNjIgMi4xNTYyNSAtNi4xNzU3ODEgMi4xNTYyNSAtNC44NzUgQyAyLjE1NjI1IC0zLjU5Mzc1IDIuNDUzMTI1IC0yLjYwOTM3NSAzLjA0Njg3NSAtMS45MjE4NzUgQyAzLjY0MDYyNSAtMS4yMzQzNzUgNC40OTIxODggLTAuODkwNjI1IDUuNjA5Mzc1IC0wLjg5MDYyNSBDIDYuMTc5Njg4IC0wLjg5MDYyNSA2LjcxODc1IC0wLjk5MjE4OCA3LjIxODc1IC0xLjIwMzEyNSBDIDcuNzE4NzUgLTEuNDEwMTU2IDguMTkxNDA2IC0xLjcyMjY1NiA4LjY0MDYyNSAtMi4xNDA2MjUgTCA4LjY0MDYyNSAtMC43NSBDIDguMTc5Njg4IC0wLjQzNzUgNy42OTE0MDYgLTAuMjAzMTI1IDcuMTcxODc1IC0wLjA0Njg3NSBDIDYuNjQ4NDM4IDAuMTA5Mzc1IDYuMTAxNTYyIDAuMTg3NSA1LjUzMTI1IDAuMTg3NSBDIDQuMDUwNzgxIDAuMTg3NSAyLjg4MjgxMiAtMC4yNjU2MjUgMi4wMzEyNSAtMS4xNzE4NzUgQyAxLjE3NTc4MSAtMi4wNzgxMjUgMC43NSAtMy4zMTI1IDAuNzUgLTQuODc1IEMgMC43NSAtNi40NDUzMTIgMS4xNzU3ODEgLTcuNjg3NSAyLjAzMTI1IC04LjU5Mzc1IEMgMi44ODI4MTIgLTkuNTA3ODEyIDQuMDUwNzgxIC05Ljk2ODc1IDUuNTMxMjUgLTkuOTY4NzUgQyA2LjExMzI4MSAtOS45Njg3NSA2LjY2NDA2MiAtOS44OTA2MjUgNy4xODc1IC05LjczNDM3NSBDIDcuNzA3MDMxIC05LjU3ODEyNSA4LjE5MTQwNiAtOS4zNDM3NSA4LjY0MDYyNSAtOS4wMzEyNSBaIE0gOC42NDA2MjUgLTkuMDMxMjUgIi8+CjwvZz4KPGcgaWQ9ImdseXBoLTAtNSI+CjxwYXRoIGQ9Ik0gMS4zMTI1IC05Ljc4MTI1IEwgMi42NDA2MjUgLTkuNzgxMjUgTCAyLjY0MDYyNSAtNS43ODEyNSBMIDcuNDUzMTI1IC01Ljc4MTI1IEwgNy40NTMxMjUgLTkuNzgxMjUgTCA4Ljc4MTI1IC05Ljc4MTI1IEwgOC43ODEyNSAwIEwgNy40NTMxMjUgMCBMIDcuNDUzMTI1IC00LjY1NjI1IEwgMi42NDA2MjUgLTQuNjU2MjUgTCAyLjY0MDYyNSAwIEwgMS4zMTI1IDAgWiBNIDEuMzEyNSAtOS43ODEyNSAiLz4KPC9nPgo8ZyBpZD0iZ2x5cGgtMC02Ij4KPHBhdGggZD0iTSAxLjMxMjUgLTkuNzgxMjUgTCAzLjA5Mzc1IC05Ljc4MTI1IEwgNy40Mzc1IC0xLjU5Mzc1IEwgNy40Mzc1IC05Ljc4MTI1IEwgOC43MTg3NSAtOS43ODEyNSBMIDguNzE4NzUgMCBMIDYuOTM3NSAwIEwgMi42MDkzNzUgLTguMTg3NSBMIDIuNjA5Mzc1IDAgTCAxLjMxMjUgMCBaIE0gMS4zMTI1IC05Ljc4MTI1ICIvPgo8L2c+CjxnIGlkPSJnbHlwaC0xLTAiPgo8cGF0aCBkPSJNIDAuNDUzMTI1IDEuNTc4MTI1IEwgMC40NTMxMjUgLTYuMzEyNSBMIDQuOTIxODc1IC02LjMxMjUgTCA0LjkyMTg3NSAxLjU3ODEyNSBaIE0gMC45NTMxMjUgMS4wNzgxMjUgTCA0LjQyMTg3NSAxLjA3ODEyNSBMIDQuNDIxODc1IC01LjgxMjUgTCAwLjk1MzEyNSAtNS44MTI1IFogTSAwLjk1MzEyNSAxLjA3ODEyNSAiLz4KPC9nPgo8ZyBpZD0iZ2x5cGgtMS0xIj4KPHBhdGggZD0iTSAzLjY0MDYyNSAtMy41MTU2MjUgQyA0LjA1NDY4OCAtMy40Mjk2ODggNC4zODI4MTIgLTMuMjQyMTg4IDQuNjI1IC0yLjk1MzEyNSBDIDQuODYzMjgxIC0yLjY3MTg3NSA0Ljk4NDM3NSAtMi4zMTY0MDYgNC45ODQzNzUgLTEuODkwNjI1IEMgNC45ODQzNzUgLTEuMjUzOTA2IDQuNzU3ODEyIC0wLjc1NzgxMiA0LjMxMjUgLTAuNDA2MjUgQyAzLjg3NSAtMC4wNTA3ODEyIDMuMjQyMTg4IDAuMTI1IDIuNDIxODc1IDAuMTI1IEMgMi4xNDg0MzggMC4xMjUgMS44NjcxODggMC4wOTc2NTYyIDEuNTc4MTI1IDAuMDQ2ODc1IEMgMS4yODUxNTYgLTAuMDAzOTA2MjUgMC45ODgyODEgLTAuMDg1OTM3NSAwLjY4NzUgLTAuMjAzMTI1IEwgMC42ODc1IC0xLjA0Njg3NSBDIDAuOTI1NzgxIC0wLjg5ODQzOCAxLjE5MTQwNiAtMC43ODkwNjIgMS40ODQzNzUgLTAuNzE4NzUgQyAxLjc3MzQzOCAtMC42NDQ1MzEgMi4wODIwMzEgLTAuNjA5Mzc1IDIuNDA2MjUgLTAuNjA5Mzc1IEMgMi45NTcwMzEgLTAuNjA5Mzc1IDMuMzc1IC0wLjcxODc1IDMuNjU2MjUgLTAuOTM3NSBDIDMuOTQ1MzEyIC0xLjE1NjI1IDQuMDkzNzUgLTEuNDcyNjU2IDQuMDkzNzUgLTEuODkwNjI1IEMgNC4wOTM3NSAtMi4yNzM0MzggMy45NTcwMzEgLTIuNTc4MTI1IDMuNjg3NSAtMi43OTY4NzUgQyAzLjQyNTc4MSAtMy4wMTU2MjUgMy4wNTQ2ODggLTMuMTI1IDIuNTc4MTI1IC0zLjEyNSBMIDEuODEyNSAtMy4xMjUgTCAxLjgxMjUgLTMuODQzNzUgTCAyLjYwOTM3NSAtMy44NDM3NSBDIDMuMDM1MTU2IC0zLjg0Mzc1IDMuMzYzMjgxIC0zLjkyOTY4OCAzLjU5Mzc1IC00LjEwOTM3NSBDIDMuODMyMDMxIC00LjI4NTE1NiAzLjk1MzEyNSAtNC41MzUxNTYgMy45NTMxMjUgLTQuODU5Mzc1IEMgMy45NTMxMjUgLTUuMTkxNDA2IDMuODMyMDMxIC01LjQ0NTMxMiAzLjU5Mzc1IC01LjYyNSBDIDMuMzUxNTYyIC01LjgxMjUgMy4wMTU2MjUgLTUuOTA2MjUgMi41NzgxMjUgLTUuOTA2MjUgQyAyLjMyODEyNSAtNS45MDYyNSAyLjA2MjUgLTUuODc4OTA2IDEuNzgxMjUgLTUuODI4MTI1IEMgMS41MDc4MTIgLTUuNzczNDM4IDEuMjA3MDMxIC01LjY5MTQwNiAwLjg3NSAtNS41NzgxMjUgTCAwLjg3NSAtNi4zNTkzNzUgQyAxLjIwNzAzMSAtNi40NTMxMjUgMS41MTk1MzEgLTYuNTE5NTMxIDEuODEyNSAtNi41NjI1IEMgMi4xMTMyODEgLTYuNjEzMjgxIDIuMzk0NTMxIC02LjY0MDYyNSAyLjY1NjI1IC02LjY0MDYyNSBDIDMuMzIwMzEyIC02LjY0MDYyNSAzLjg0NzY1NiAtNi40ODgyODEgNC4yMzQzNzUgLTYuMTg3NSBDIDQuNjI4OTA2IC01Ljg4MjgxMiA0LjgyODEyNSAtNS40NzI2NTYgNC44MjgxMjUgLTQuOTUzMTI1IEMgNC44MjgxMjUgLTQuNTg1OTM4IDQuNzIyNjU2IC00LjI4MTI1IDQuNTE1NjI1IC00LjAzMTI1IEMgNC4zMDQ2ODggLTMuNzgxMjUgNC4wMTU2MjUgLTMuNjA5Mzc1IDMuNjQwNjI1IC0zLjUxNTYyNSBaIE0gMy42NDA2MjUgLTMuNTE1NjI1ICIvPgo8L2c+CjwvZz4KPC9kZWZzPgo8cGF0aCBmaWxsPSJub25lIiBzdHJva2Utd2lkdGg9IjAuMDMzMzMzMyIgc3Ryb2tlLWxpbmVjYXA9ImJ1dHQiIHN0cm9rZS1saW5lam9pbj0ibWl0ZXIiIHN0cm9rZT0icmdiKDAlLCAwJSwgMCUpIiBzdHJva2Utb3BhY2l0eT0iMSIgc3Ryb2tlLW1pdGVybGltaXQ9IjEwIiBkPSJNIDQuOTIxMjM2IDEuMzI1MTk1IEwgNC41OTczNDIgMS41NzQwMjIgIiB0cmFuc2Zvcm09Im1hdHJpeCgzMy41NjM4MDgsIDAsIDAsIDMzLjU2MzgwOCwgMjEuMzk0ODk4LCA5Ni4wNDA5MzQpIi8+CjxwYXRoIGZpbGw9Im5vbmUiIHN0cm9rZS13aWR0aD0iMC4wMzMzMzMzIiBzdHJva2UtbGluZWNhcD0iYnV0dCIgc3Ryb2tlLWxpbmVqb2luPSJtaXRlciIgc3Ryb2tlPSJyZ2IoMCUsIDAlLCAwJSkiIHN0cm9rZS1vcGFjaXR5PSIxIiBzdHJva2UtbWl0ZXJsaW1pdD0iMTAiIGQ9Ik0gNS4zODI2OTQgMS4zMjUwNzkgTCA1LjY4NjkxOCAxLjU1ODQyNiAiIHRyYW5zZm9ybT0ibWF0cml4KDMzLjU2MzgwOCwgMCwgMCwgMzMuNTYzODA4LCAyMS4zOTQ4OTgsIDk2LjA0MDkzNCkiLz4KPHBhdGggZmlsbD0ibm9uZSIgc3Ryb2tlLXdpZHRoPSIwLjAzMzMzMzMiIHN0cm9rZS1saW5lY2FwPSJidXR0IiBzdHJva2UtbGluZWpvaW49Im1pdGVyIiBzdHJva2U9InJnYigwJSwgMCUsIDAlKSIgc3Ryb2tlLW9wYWNpdHk9IjEiIHN0cm9rZS1taXRlcmxpbWl0PSIxMCIgZD0iTSA1LjQxNTUxNCAwLjk4NDA3NyBMIDUuNjY1ODUzIDAuNzExMDQzICIgdHJhbnNmb3JtPSJtYXRyaXgoMzMuNTYzODA4LCAwLCAwLCAzMy41NjM4MDgsIDIxLjM5NDg5OCwgOTYuMDQwOTM0KSIvPgo8cGF0aCBmaWxsPSJub25lIiBzdHJva2Utd2lkdGg9IjAuMDMzMzMzMyIgc3Ryb2tlLWxpbmVjYXA9ImJ1dHQiIHN0cm9rZS1saW5lam9pbj0ibWl0ZXIiIHN0cm9rZT0icmdiKDAlLCAwJSwgMCUpIiBzdHJva2Utb3BhY2l0eT0iMSIgc3Ryb2tlLW1pdGVybGltaXQ9IjEwIiBkPSJNIDUuMjkyNjEzIDAuODcxNDE5IEwgNS41NDMwNjkgMC41OTgzODUgIiB0cmFuc2Zvcm09Im1hdHJpeCgzMy41NjM4MDgsIDAsIDAsIDMzLjU2MzgwOCwgMjEuMzk0ODk4LCA5Ni4wNDA5MzQpIi8+CjxwYXRoIGZpbGw9Im5vbmUiIHN0cm9rZS13aWR0aD0iMC4wMzMzMzMzIiBzdHJva2UtbGluZWNhcD0iYnV0dCIgc3Ryb2tlLWxpbmVqb2luPSJtaXRlciIgc3Ryb2tlPSJyZ2IoMCUsIDAlLCAwJSkiIHN0cm9rZS1vcGFjaXR5PSIxIiBzdHJva2UtbWl0ZXJsaW1pdD0iMTAiIGQ9Ik0gNC45NDk5ODIgMC45Mjc2MzIgTCA0LjY5MjE5NCAwLjY0NjEwMiAiIHRyYW5zZm9ybT0ibWF0cml4KDMzLjU2MzgwOCwgMCwgMCwgMzMuNTYzODA4LCAyMS4zOTQ4OTgsIDk2LjA0MDkzNCkiLz4KPHBhdGggZmlsbD0ibm9uZSIgc3Ryb2tlLXdpZHRoPSIwLjAzMzMzMzMiIHN0cm9rZS1saW5lY2FwPSJidXR0IiBzdHJva2UtbGluZWpvaW49Im1pdGVyIiBzdHJva2U9InJnYigwJSwgMCUsIDAlKSIgc3Ryb2tlLW9wYWNpdHk9IjEiIHN0cm9rZS1taXRlcmxpbWl0PSIxMCIgZD0iTSA0LjExMjg0MSAxLjY1NTQ5IEwgMy40MjQ2NjkgMS4zNzA0NjggIiB0cmFuc2Zvcm09Im1hdHJpeCgzMy41NjM4MDgsIDAsIDAsIDMzLjU2MzgwOCwgMjEuMzk0ODk4LCA5Ni4wNDA5MzQpIi8+CjxwYXRoIGZpbGw9Im5vbmUiIHN0cm9rZS13aWR0aD0iMC4wMzMzMzMzIiBzdHJva2UtbGluZWNhcD0iYnV0dCIgc3Ryb2tlLWxpbmVqb2luPSJtaXRlciIgc3Ryb2tlPSJyZ2IoMCUsIDAlLCAwJSkiIHN0cm9rZS1vcGFjaXR5PSIxIiBzdHJva2UtbWl0ZXJsaW1pdD0iMTAiIGQ9Ik0gNi4yMjQxNDEgMS42NDE5ODkgTCA2Ljg3ODkxMSAxLjM3MDQ2OCAiIHRyYW5zZm9ybT0ibWF0cml4KDMzLjU2MzgwOCwgMCwgMCwgMzMuNTYzODA4LCAyMS4zOTQ4OTgsIDk2LjA0MDkzNCkiLz4KPHBhdGggZmlsbD0ibm9uZSIgc3Ryb2tlLXdpZHRoPSIwLjAzMzMzMzMiIHN0cm9rZS1saW5lY2FwPSJidXR0IiBzdHJva2UtbGluZWpvaW49Im1pdGVyIiBzdHJva2U9InJnYigwJSwgMCUsIDAlKSIgc3Ryb2tlLW9wYWNpdHk9IjEiIHN0cm9rZS1taXRlcmxpbWl0PSIxMCIgZD0iTSAzLjQ0MTE5NiAxLjM2ODI1NyBMIDIuNjMxODcgMS45ODgyMjggIiB0cmFuc2Zvcm09Im1hdHJpeCgzMy41NjM4MDgsIDAsIDAsIDMzLjU2MzgwOCwgMjEuMzk0ODk4LCA5Ni4wNDA5MzQpIi8+CjxwYXRoIGZpbGw9Im5vbmUiIHN0cm9rZS13aWR0aD0iMC4wMzMzMzMzIiBzdHJva2UtbGluZWNhcD0iYnV0dCIgc3Ryb2tlLWxpbmVqb2luPSJtaXRlciIgc3Ryb2tlPSJyZ2IoMCUsIDAlLCAwJSkiIHN0cm9rZS1vcGFjaXR5PSIxIiBzdHJva2UtbWl0ZXJsaW1pdD0iMTAiIGQ9Ik0gNi44NjIzODQgMS4zNjgyNTcgTCA3LjQxNTA4NiAxLjc5MjAwNyAiIHRyYW5zZm9ybT0ibWF0cml4KDMzLjU2MzgwOCwgMCwgMCwgMzMuNTYzODA4LCAyMS4zOTQ4OTgsIDk2LjA0MDkzNCkiLz4KPHBhdGggZmlsbD0ibm9uZSIgc3Ryb2tlLXdpZHRoPSIwLjAzMzMzMzMiIHN0cm9rZS1saW5lY2FwPSJidXR0IiBzdHJva2UtbGluZWpvaW49Im1pdGVyIiBzdHJva2U9InJnYigwJSwgMCUsIDAlKSIgc3Ryb2tlLW9wYWNpdHk9IjEiIHN0cm9rZS1taXRlcmxpbWl0PSIxMCIgZD0iTSAyLjY0ODI4IDEuOTg2MDE3IEwgMS45Njg4MzcgMS43MDUwNjkgIiB0cmFuc2Zvcm09Im1hdHJpeCgzMy41NjM4MDgsIDAsIDAsIDMzLjU2MzgwOCwgMjEuMzk0ODk4LCA5Ni4wNDA5MzQpIi8+CjxwYXRoIGZpbGw9Im5vbmUiIHN0cm9rZS13aWR0aD0iMC4wMzMzMzMzIiBzdHJva2UtbGluZWNhcD0iYnV0dCIgc3Ryb2tlLWxpbmVqb2luPSJtaXRlciIgc3Ryb2tlPSJyZ2IoMCUsIDAlLCAwJSkiIHN0cm9rZS1vcGFjaXR5PSIxIiBzdHJva2UtbWl0ZXJsaW1pdD0iMTAiIGQ9Ik0gMS40NzQzMjYgMS43ODUyNTYgTCAwLjkyMzEzNyAyLjIwODQyNCAiIHRyYW5zZm9ybT0ibWF0cml4KDMzLjU2MzgwOCwgMCwgMCwgMzMuNTYzODA4LCAyMS4zOTQ4OTgsIDk2LjA0MDkzNCkiLz4KPHBhdGggZmlsbD0ibm9uZSIgc3Ryb2tlLXdpZHRoPSIwLjAzMzMzMzMiIHN0cm9rZS1saW5lY2FwPSJidXR0IiBzdHJva2UtbGluZWpvaW49Im1pdGVyIiBzdHJva2U9InJnYigwJSwgMCUsIDAlKSIgc3Ryb2tlLW9wYWNpdHk9IjEiIHN0cm9rZS1taXRlcmxpbWl0PSIxMCIgZD0iTSAxLjY4MDA5MSAxLjMzMjA2MiBMIDEuNTg1MjM5IDAuNjA5MDkyICIgdHJhbnNmb3JtPSJtYXRyaXgoMzMuNTYzODA4LCAwLCAwLCAzMy41NjM4MDgsIDIxLjM5NDg5OCwgOTYuMDQwOTM0KSIvPgo8cGF0aCBmaWxsPSJub25lIiBzdHJva2Utd2lkdGg9IjAuMDMzMzMzMyIgc3Ryb2tlLWxpbmVjYXA9ImJ1dHQiIHN0cm9rZS1saW5lam9pbj0ibWl0ZXIiIHN0cm9rZT0icmdiKDAlLCAwJSwgMCUpIiBzdHJva2Utb3BhY2l0eT0iMSIgc3Ryb2tlLW1pdGVybGltaXQ9IjEwIiBkPSJNIDAuOTIzMTM3IDIuMjA4NDI0IEwgMS4wMTc5ODkgMi45MjU2OTEgIiB0cmFuc2Zvcm09Im1hdHJpeCgzMy41NjM4MDgsIDAsIDAsIDMzLjU2MzgwOCwgMjEuMzk0ODk4LCA5Ni4wNDA5MzQpIi8+CjxwYXRoIGZpbGw9Im5vbmUiIHN0cm9rZS13aWR0aD0iMC4wMzMzMzMzIiBzdHJva2UtbGluZWNhcD0iYnV0dCIgc3Ryb2tlLWxpbmVqb2luPSJtaXRlciIgc3Ryb2tlPSJyZ2IoMCUsIDAlLCAwJSkiIHN0cm9rZS1vcGFjaXR5PSIxIiBzdHJva2UtbWl0ZXJsaW1pdD0iMTAiIGQ9Ik0gMC45MjMxMzcgMi4yMDg0MjQgTCAwLjI2MDgwMyAxLjkzMzY0NCAiIHRyYW5zZm9ybT0ibWF0cml4KDMzLjU2MzgwOCwgMCwgMCwgMzMuNTYzODA4LCAyMS4zOTQ4OTgsIDk2LjA0MDkzNCkiLz4KPHBhdGggZmlsbD0ibm9uZSIgc3Ryb2tlLXdpZHRoPSIwLjAzMzMzMzMiIHN0cm9rZS1saW5lY2FwPSJidXR0IiBzdHJva2UtbGluZWpvaW49Im1pdGVyIiBzdHJva2U9InJnYigwJSwgMCUsIDAlKSIgc3Ryb2tlLW9wYWNpdHk9IjEiIHN0cm9rZS1taXRlcmxpbWl0PSIxMCIgZD0iTSAxLjU4NTIzOSAwLjYwOTA5MiBMIDIuMTMxMTkxIDAuMTkwMzQ3ICIgdHJhbnNmb3JtPSJtYXRyaXgoMzMuNTYzODA4LCAwLCAwLCAzMy41NjM4MDgsIDIxLjM5NDg5OCwgOTYuMDQwOTM0KSIvPgo8cGF0aCBmaWxsPSJub25lIiBzdHJva2Utd2lkdGg9IjAuMDMzMzMzMyIgc3Ryb2tlLWxpbmVjYXA9ImJ1dHQiIHN0cm9rZS1saW5lam9pbj0ibWl0ZXIiIHN0cm9rZT0icmdiKDAlLCAwJSwgMCUpIiBzdHJva2Utb3BhY2l0eT0iMSIgc3Ryb2tlLW1pdGVybGltaXQ9IjEwIiBkPSJNIDEuNTg1MjM5IDAuNjA5MDkyIEwgMC45MjMwMjEgMC4zMzQxOTYgIiB0cmFuc2Zvcm09Im1hdHJpeCgzMy41NjM4MDgsIDAsIDAsIDMzLjU2MzgwOCwgMjEuMzk0ODk4LCA5Ni4wNDA5MzQpIi8+CjxnIGZpbGw9InJnYig4NC4zODgxOTElLCA0Mi4xOTQwOTUlLCAwJSkiIGZpbGwtb3BhY2l0eT0iMSI+Cjx1c2UgeGxpbms6aHJlZj0iI2dseXBoLTAtMSIgeD0iMTg5LjgzNTkzOCIgeT0iMTM5LjQ2ODc1Ii8+CjwvZz4KPGcgZmlsbD0icmdiKDY1LjY3NzY0MyUsIDUwLjIyNDA3OCUsIDcuNzI2NzgyJSkiIGZpbGwtb3BhY2l0eT0iMSI+Cjx1c2UgeGxpbms6aHJlZj0iI2dseXBoLTAtMiIgeD0iMTYzLjM1OTM3NSIgeT0iMTU5LjkxMDE1NiIvPgo8L2c+CjxnIGZpbGw9InJnYigxMDAlLCA1LjElLCA1LjElKSIgZmlsbC1vcGFjaXR5PSIxIj4KPHVzZSB4bGluazpocmVmPSIjZ2x5cGgtMC0zIiB4PSIyMTUuNjc5Njg4IiB5PSIxNTkuOTEwMTU2Ii8+CjwvZz4KPGcgZmlsbD0icmdiKDEwMCUsIDUuMSUsIDUuMSUpIiBmaWxsLW9wYWNpdHk9IjEiPgo8dXNlIHhsaW5rOmhyZWY9IiNnbHlwaC0wLTMiIHg9IjIxMS43MTg3NSIgeT0iMTE0LjcyMjY1NiIvPgo8L2c+CjxnIGZpbGw9InJnYigwJSwgMCUsIDAlKSIgZmlsbC1vcGFjaXR5PSIxIj4KPHVzZSB4bGluazpocmVmPSIjZ2x5cGgtMC00IiB4PSIxNjYuOTQ5MjE5IiB5PSIxMTQuNzIyNjU2Ii8+CjwvZz4KPGcgZmlsbD0icmdiKDAlLCAwJSwgMCUpIiBmaWxsLW9wYWNpdHk9IjEiPgo8dXNlIHhsaW5rOmhyZWY9IiNnbHlwaC0wLTUiIHg9IjE1Mi4zOTQ1MzEiIHk9IjExNC41NDY4NzUiLz4KPC9nPgo8ZyBmaWxsPSJyZ2IoMCUsIDAlLCAwJSkiIGZpbGwtb3BhY2l0eT0iMSI+Cjx1c2UgeGxpbms6aHJlZj0iI2dseXBoLTEtMSIgeD0iMTYxLjYwOTM3NSIgeT0iMTE1LjMxNjQwNiIvPgo8L2c+CjxnIGZpbGw9InJnYigwJSwgMCUsIDAlKSIgZmlsbC1vcGFjaXR5PSIxIj4KPHVzZSB4bGluazpocmVmPSIjZ2x5cGgtMC00IiB4PSIyNzMuOTAyMzQ0IiB5PSIxNjcuNDYwOTM4Ii8+CjwvZz4KPGcgZmlsbD0icmdiKDAlLCAwJSwgMCUpIiBmaWxsLW9wYWNpdHk9IjEiPgo8dXNlIHhsaW5rOmhyZWY9IiNnbHlwaC0wLTUiIHg9IjI4Mi4zNTE1NjIiIHk9IjE2Ny4yODUxNTYiLz4KPC9nPgo8ZyBmaWxsPSJyZ2IoMCUsIDAlLCAwJSkiIGZpbGwtb3BhY2l0eT0iMSI+Cjx1c2UgeGxpbms6aHJlZj0iI2dseXBoLTEtMSIgeD0iMjkxLjU3MDMxMiIgeT0iMTY4LjA1ODU5NCIvPgo8L2c+CjxnIGZpbGw9InJnYigxOSUsIDMxJSwgOTcuMDAwMDAzJSkiIGZpbGwtb3BhY2l0eT0iMSI+Cjx1c2UgeGxpbms6aHJlZj0iI2dseXBoLTAtNiIgeD0iNzMuOTQ1MzEyIiB5PSIxNTQuNjQ0NTMxIi8+CjwvZz4KPGcgZmlsbD0icmdiKDAlLCAwJSwgMCUpIiBmaWxsLW9wYWNpdHk9IjEiPgo8dXNlIHhsaW5rOmhyZWY9IiNnbHlwaC0wLTQiIHg9IjUyLjA3NDIxOSIgeT0iMjA4LjMxNjQwNiIvPgo8L2c+CjxnIGZpbGw9InJnYigwJSwgMCUsIDAlKSIgZmlsbC1vcGFjaXR5PSIxIj4KPHVzZSB4bGluazpocmVmPSIjZ2x5cGgtMC01IiB4PSI2MC41MjM0MzgiIHk9IjIwOC4xNDA2MjUiLz4KPC9nPgo8ZyBmaWxsPSJyZ2IoMCUsIDAlLCAwJSkiIGZpbGwtb3BhY2l0eT0iMSI+Cjx1c2UgeGxpbms6aHJlZj0iI2dseXBoLTEtMSIgeD0iNjkuNzM4MjgxIiB5PSIyMDguOTEwMTU2Ii8+CjwvZz4KPGcgZmlsbD0icmdiKDAlLCAwJSwgMCUpIiBmaWxsLW9wYWNpdHk9IjEiPgo8dXNlIHhsaW5rOmhyZWY9IiNnbHlwaC0wLTQiIHg9IjE2LjY5MTQwNiIgeT0iMTYyLjE5NTMxMiIvPgo8L2c+CjxnIGZpbGw9InJnYigwJSwgMCUsIDAlKSIgZmlsbC1vcGFjaXR5PSIxIj4KPHVzZSB4bGluazpocmVmPSIjZ2x5cGgtMC01IiB4PSIyLjEzNjcxOSIgeT0iMTYyLjAxOTUzMSIvPgo8L2c+CjxnIGZpbGw9InJnYigwJSwgMCUsIDAlKSIgZmlsbC1vcGFjaXR5PSIxIj4KPHVzZSB4bGluazpocmVmPSIjZ2x5cGgtMS0xIiB4PSIxMS4zNTE1NjIiIHk9IjE2Mi43ODkwNjIiLz4KPC9nPgo8ZyBmaWxsPSJyZ2IoMCUsIDAlLCAwJSkiIGZpbGwtb3BhY2l0eT0iMSI+Cjx1c2UgeGxpbms6aHJlZj0iI2dseXBoLTAtNCIgeD0iOTYuNTU0Njg4IiB5PSIxMDAuOTI1NzgxIi8+CjwvZz4KPGcgZmlsbD0icmdiKDAlLCAwJSwgMCUpIiBmaWxsLW9wYWNpdHk9IjEiPgo8dXNlIHhsaW5rOmhyZWY9IiNnbHlwaC0wLTUiIHg9IjEwNS4wMDM5MDYiIHk9IjEwMC43NSIvPgo8L2c+CjxnIGZpbGw9InJnYigwJSwgMCUsIDAlKSIgZmlsbC1vcGFjaXR5PSIxIj4KPHVzZSB4bGluazpocmVmPSIjZ2x5cGgtMS0xIiB4PSIxMTQuMjE4NzUiIHk9IjEwMS41MjM0MzgiLz4KPC9nPgo8ZyBmaWxsPSJyZ2IoMCUsIDAlLCAwJSkiIGZpbGwtb3BhY2l0eT0iMSI+Cjx1c2UgeGxpbms6aHJlZj0iI2dseXBoLTAtNCIgeD0iMzguOTE3OTY5IiB5PSIxMDguNTExNzE5Ii8+CjwvZz4KPGcgZmlsbD0icmdiKDAlLCAwJSwgMCUpIiBmaWxsLW9wYWNpdHk9IjEiPgo8dXNlIHhsaW5rOmhyZWY9IiNnbHlwaC0wLTUiIHg9IjI0LjM2MzI4MSIgeT0iMTA4LjMzNTkzOCIvPgo8L2c+CjxnIGZpbGw9InJnYigwJSwgMCUsIDAlKSIgZmlsbC1vcGFjaXR5PSIxIj4KPHVzZSB4bGluazpocmVmPSIjZ2x5cGgtMS0xIiB4PSIzMy41NzgxMjUiIHk9IjEwOS4xMDU0NjkiLz4KPC9nPgo8L3N2Zz4K)