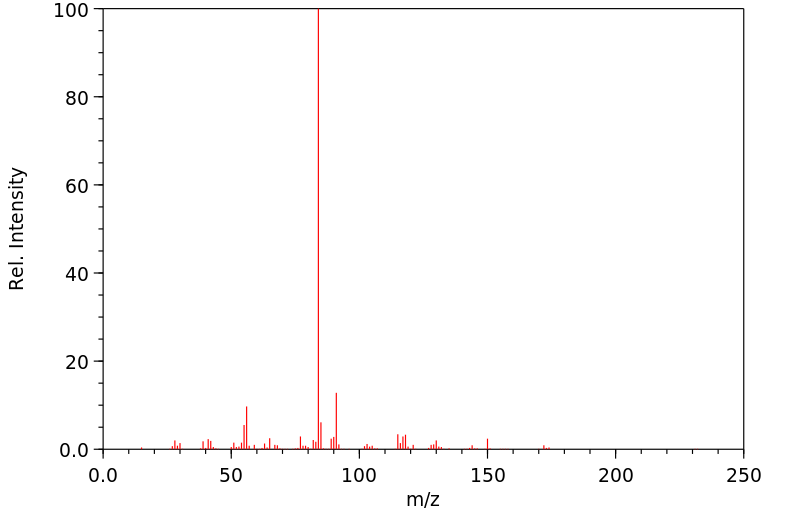
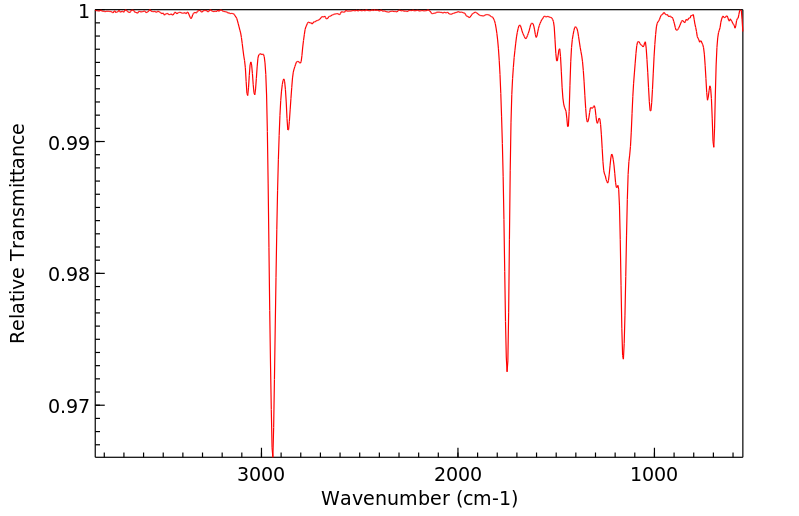
Methylphenidate is hepatically metabolized. More specifically, it is rapidly and extensively metabolized by carboxylesterase CES1A1. Via this enzyme, methylphenidate undergoes de-esterification to ritalinic acid (a-phenyl-2-piperidine acetic acid, PPAA), which has little to no pharmacologic activity.
In humans, methylphenidate is metabolized primarily via deesterification to alpha-phenylpiperidine acetic acid (PPA, ritalinic acid). The metabolite has little or no pharmacologic activity.
Methylphenidate is hepatically metabolized. More specifically, it is rapidly and extensively metabolized by carboxylesterase CES1A1. Via this enzyme, methylphenidate undergoes de-esterification to ritalinic acid (a-phenyl-2-piperidine acetic acid, PPAA), which has little to no pharmacologic activity.
Route of Elimination: After oral administration of an immediate release formulation of methylphenidate, 78%-97% of the dose is excreted in the urine and 1%-3% in the feces in the form of metabolites within 48-96 hours. Only small quantities (<1%) of unchanged methylphenidate appear in the urine. Most of the dose is excreted in the urine as ritalinic acid (60%-86%), the remainder being accounted for by minor metabolites.
Half Life: d-methylphenidate = 3-4 hours;
l-methylphenidate = 1-3 hours;
Ritalinic acid = 3-4 hours;
epatic, methylphenidate is metabolized primarily by de-esterification to ritalinic acid (α-phenyl-2-piperidine acetic acid, PPAA), which has little to no pharmacologic activity.
Route of Elimination: Renal
Half Life: 2-4 hours
IDENTIFICATION AND USE: Methylphenidate Hydrochloride is a psychostimulant used to treat different behavioral disorders. Indications: Used to treat narcolepsy and hyperkinetic states in children (as an adjunct to psychological, educational and social measures) for amphetamine, dextroamphetamine and ethylphenidate. It is misused for performance enhancement and relief of fatigue. Abuse either orally or by injection is extremely common. HUMAN EXPOSURE AND TOXICITY: Main risks include: Acute central nervous system (CNS) stimulation, cardiotoxicity causing tachycardia, arrhythmias, hypertension and cardiovascular collapse. High risk of dependency and abuse. Cardiovascular effects include: palpitation, chest pain, tachycardia, arrhythmias and hypertension; cardiovascular collapse can occur in severe poisoning. Myocardial ischemia, infarction and ventricular dysfunction are described. CNS effects include: stimulation of CNS, tremor, restlessness, agitation, insomnia, increased motor activity, headache, convulsions, coma and hyperreflexia. Stroke and cerebral vasculitis have been observed. Gastrointestinal effects include: vomiting, diarrhea and cramps. Genitourinary effects include: increased bladder sphincter tone which may cause dysuria, hesitancy and acute urinary retention. Renal failure can occur secondary to dehydration or rhabdomyolysis. Renal ischemia may be noted. Transient hyperthyroxinemia may be noted. Increased metabolic and muscular activity may result in hyperventilation and hyperthermia. Weight loss is common with chronic use. Hypo- and hyperkalemia have been reported. Dehydration is common. Fasciculations and rigidity may be noted. Rhabdomyolysis is an important consequence of severe poisoning. Agitation, confusion, mood elevation, increased wakefulness, talkativeness, irritability and panic attacks are typical. Chronic abuse can cause delusions and paranoia. A withdrawal syndrome occurs after abrupt cessation following chronic use. Appears to exert most or all of its effect in the CNS by causing release of biogenic amines, especially norepinephrine and dopamine, from storage sites in nerve terminals. It may also slow down catecholamine metabolism by inhibiting monoamine oxidase. Does not appear to be a human teratogen. Mild withdrawal symptoms may be observed in the newborn, but the few studies of infant follow-up have not shown long-term sequelae, although more studies of this nature are needed. Illicit maternal use or abuse presents a significant risk to the fetus and newborn, including intrauterine growth retardation, premature delivery and the potential for increased maternal, fetal and neonatal morbidity. Cerebral injuries occurring in newborns exposed in utero appear to be directly related to the vasoconstrictive properties of the drug. ANIMAL STUDIES: In 2 years feed studies, there was no evidence of carcinogenic activity of methylphenidate hydrochloride in male or female rats receiving 100, 500 or 1,000 ppm. There was some evidence of carcinogenic activity of methylphenidate hydrochloride in male or female mice based on the occurrence of hepatocellular neoplasms. In the squirrel monkey methylphenidate produced modest increases in masked auditory thresholds.
Methylphenidate blocks dopamine uptake in central adrenergic neurons by blocking dopamine transport or carrier proteins. Methylphenidate acts at the brain stem arousal system and the cerebral cortex and causes increased sympathomimetic activity in the central nervous system. Alteration of serotonergic pathways via changes in dopamine transport may result.
Methylphenidate blocks dopamine uptake in central adrenergic neurons by blocking dopamine transport or carrier proteins. Methylphenidate acts at the brain stem arousal system and the cerebral cortex and causes increased sympathomimetic activity in the central nervous system.
Methylphenidate is a catecholamine reuptake inhibitor that indirectly increases catecholaminergic neurotransmission by inhibiting the dopamine transporter (DAT) and norepinephrine transporter (NET), which are responsible for clearing catecholamines from the synapse, particularly in the striatum and meso-limbic system.
In prelicensure clinical trials, methylphenidate was not associated with serum aminotransferase elevations or instances of hepatic injury, but reports of enzyme elevations were received by the sponsor and appeared in publications after it was marketed. The elevations were transient, mild-to-moderate in severity and rarely associated with jaundice or symptoms. In addition, there have been several case reports of marked serum enzyme elevations with jaundice and clinically apparent acute liver injury attributed to methylphenidate given intravenously. These cases occurred largely after illicit intravenous use of methylphenidate and frequently in patients with underlying chronic hepatitis C. The onset was generally soon after initiation of therapy. The pattern of liver enzyme elevations was hepatocellular and the clinical phenotype typical of acute hepatic necrosis with rapid onset and rapid recovery. Immunoallergic features were uncommon. A single case associated with low levels of autoantibodies has been published. Some cases have been severe and at least one fatality after intravenous abuse has been reported. Recurrence upon rechallenge was been documented in one instance (Case 1).
Concerta®: Methylphenidate is readily absorbed. Following oral administration of Concerta, plasma methylhphenidate concentrations reach an initial maximum at about 1 hour followed by gradual ascending concentrations over the next 5-9 hours. Mean times to reach peak plasma concentrations across all doses of Concerta occurred between 6-10 hours. Once daily dosing minimizes the fluctuations between peak and trough concentrations associated with multiple doses of immediate-release methylphenidate treatments. Depending on the doses provided, Cmax was found to range from 6.0-15.0ng/mL, Tmax ranged from 8.1-9.4h, and AUC ranged from 50.4-121.5 ng·h/mL in children. When provided as Concerta®, methylphenidate is released through the patented Osmotic Controlled-Release Oral Delivery (OROS) system where 22% of the dose is provided as an immediate release and 78% is provided through a gradual release. OROS is comprised of an osmotically active trilayer core surrounded by a semipermeable membrane with an immediate-release drug overcoat. Within an aqueous environment, such as the stomach, the drug overcoat, which consists of 22% of the dose, dissolves within one hour, providing an initial immediate-release formulation of methylphenidate. Water then permeates through the membrane into the tablet core where the osmotically active polymer excipients expand, allowing methylphenidate to release slowly through the orifice over a period of 6-7 hours. Concerta also provides a sustained 10-12 hour effect, allowing for once-daily dosing. Biphentin®: Methylphenidate is rapidly and extensively absorbed following oral administration, with peak blood levels obtained in 1-3 hours. When provided as Biphentin®, methylphenidate is released through a multi-layer release delivery system (MLRTM) where 40% of the dose is provided as an immediate release and 60% is provided through a gradual release. Biphentin was designed to be an alternative to separate doses of immediate-release (IR) methylphenidate by providing a biphasic concentration-time profile when given as a single dose. The MLRTM release system allows for a sustained effect for 10-12 hours, allowing for once-daily dosing that covers the major times that ADHD impairment might occur (such as school, homework periods, during the workday, etc). Methylphenidate (immediate release): Methylphenidate hydrochloride is rapidly and extensively absorbed from the tablets following oral administration; however, owing to extensive first-pass metabolism, bioavailability is low (approx. 30%) and large individual differences exist (11-52%). In one study, the administration of methylphenidate hydrochloride with food accelerated absorption but had no effect on the amount absorbed. Peak plasma concentrations of 10.8 and 7.8 ng/mL were observed, on average, 2 hours after administration of 0.30 mg/kg in children and adults, respectively. Peak plasma concentrations showed marked variability between subjects. Both the area under the concentration-time curve (AUC), and the peak plasma concentrations (Cmax) showed dose-proportionality.
After oral administration of an immediate release formulation of methylphenidate, 78%-97% of the dose is excreted in the urine and 1%-3% in the feces in the form of metabolites within 48-96 hours. Only small quantities (<1%) of unchanged methylphenidate appear in the urine. Most of the dose is excreted in the urine as ritalinic acid (60%-86%), the remainder being accounted for by minor metabolites.
Concerta: Plasma methylphenidate concentrations in adults decline bi-exponentially following oral administration. Biphentin: The apparent distribution volume of methylphenidate in children is approximately 20 L/kg, with substantial variability (11 to 33 L/kg). Methylphenidate (immediate release): The apparent distribution volume of methylphenidate in children was approximately 20 L/kg, with substantial variability (11-33 L/kg). The volume of distribution after an intravenous dose (Vss) is 2.23 L/kg for the racemate in healthy adult volunteers.
The apparent mean systemic clearance after an oral dose is 10.2 and 10.5 L/h/kg in children and adults, respectively for a 0.3 mg/kg dose, and 0.565 L/h/kg after an intravenous dose of the racemate in healthy adult volunteers.
Methylphenidate HCl Oral Solution is readily absorbed. Following oral administration of Methylphenidate HCl Oral Solution, peak plasma methylphenidate concentrations are achieved at 1 to 2 hours.
[EN] METHYL OXAZOLE OREXIN RECEPTOR ANTAGONISTS<br/>[FR] MÉTHYLOXAZOLES ANTAGONISTES DU RÉCEPTEUR DE L'OREXINE
申请人:MERCK SHARP & DOHME
公开号:WO2016089721A1
公开(公告)日:2016-06-09
The present invention is directed to methyl oxazole compounds which are antagonists of orexin receptors. The present invention is also directed to uses of the compounds described herein in the potential treatment or prevention of neurological and psychiatric disorders and diseases in which orexin receptors are involved. The present invention is also directed to compositions comprising these compounds. The present invention is also directed to uses of these compositions in the potential prevention or treatment of such diseases in which orexin receptors are involved.
[EN] A CONJUGATE OF A CYTOTOXIC AGENT TO A CELL BINDING MOLECULE WITH BRANCHED LINKERS<br/>[FR] CONJUGUÉ D'UN AGENT CYTOTOXIQUE À UNE MOLÉCULE DE LIAISON CELLULAIRE AVEC DES LIEURS RAMIFIÉS
申请人:HANGZHOU DAC BIOTECH CO LTD
公开号:WO2020257998A1
公开(公告)日:2020-12-30
Provided is a conjugation of cytotoxic drug to a cell-binding molecule with a side-chain linker. It provides side-chain linkage methods of making a conjugate of a cytotoxic molecule to a cell-binding ligand, as well as methods of using the conjugate in targeted treatment of cancer, infection and immunological disorders.
[EN] CROSS-LINKED PYRROLOBENZODIAZEPINE DIMER (PBD) DERIVATIVE AND ITS CONJUGATES<br/>[FR] DÉRIVÉ DE DIMÈRE DE PYRROLOBENZODIAZÉPINE RÉTICULÉ (PBD) ET SES CONJUGUÉS
申请人:HANGZHOU DAC BIOTECH CO LTD
公开号:WO2020006722A1
公开(公告)日:2020-01-09
A novel cross-linked cytotoxic agents, pyrrolobenzo-diazepine dimer (PBD) derivatives, and their conjugates to a cell-binding molecule, a method for preparation of the conjugates and the therapeutic use of the conjugates.
[EN] IMIDAZOLIUM REAGENT FOR MASS SPECTROMETRY<br/>[FR] RÉACTIF D'IMIDAZOLIUM POUR SPECTROMÉTRIE DE MASSE
申请人:HOFFMANN LA ROCHE
公开号:WO2021234004A1
公开(公告)日:2021-11-25
The present invention relates to compounds which are suitable to be used in mass spectrometry as well as methods of mass spectrometric determination of analyte molecules using said compounds.