
5-(4-aminophenyl)-1,3-thiazol-2-amine | 90349-87-4
中文名称
——
中文别名
——
英文名称
5-(4-aminophenyl)-1,3-thiazol-2-amine
英文别名
5-(4-aminophenyl)-2-amino-1,3-thiazole;2-Amino-5-p-aminophenylthiazole;5-(4-amino-phenyl)-thiazol-2-ylamine;5-(p-Aminophenyl)-2-thiazolamine

CAS
90349-87-4
化学式
C9H9N3S
mdl
——
分子量
191.257
InChiKey
CGEBKMQTSUAAMN-UHFFFAOYSA-N
BEILSTEIN
——
EINECS
——
-
物化性质
-
计算性质
-
ADMET
-
安全信息
-
SDS
-
制备方法与用途
-
上下游信息
-
文献信息
-
表征谱图
-
同类化合物
-
相关功能分类
-
相关结构分类
物化性质
-
熔点:197 °C
-
沸点:417.1±20.0 °C(Predicted)
-
密度:1.347±0.06 g/cm3(Predicted)
计算性质
-
辛醇/水分配系数(LogP):1.6
-
重原子数:13
-
可旋转键数:1
-
环数:2.0
-
sp3杂化的碳原子比例:0.0
-
拓扑面积:93.2
-
氢给体数:2
-
氢受体数:4
安全信息
-
危险性防范说明:P261,P264,P270,P271,P280,P301+P312,P302+P352,P304+P340,P305+P351+P338,P330,P332+P313,P337+P313,P362,P403+P233,P405,P501
-
危险性描述:H302,H315,H319,H335
-
储存条件:存储温度应保持在2-8°C,并请避免光线直射。
上下游信息
-
下游产品
中文名称 英文名称 CAS号 化学式 分子量 —— N-[5-(4-aminophenyl)-1,3-thiazol-2-yl]-2-(benzylamino)acetamide 1444302-73-1 C18H18N4OS 338.433
反应信息
-
作为反应物:描述:5-(4-aminophenyl)-1,3-thiazol-2-amine 在 吡啶 、 sodium hydride 作用下, 以 四氢呋喃 、 mineral oil 为溶剂, 反应 8.0h, 生成 C31H36N8O3S参考文献:名称:Identification of a potent 5-phenyl-thiazol-2-ylamine-based inhibitor of FLT3 with activity against drug resistance-conferring point mutations摘要:Numerous FLT3 inhibitors have been explored as a viable therapy for the treatment of acute myeloid leukemia (AML). However, clinical data have been underwhelming due to incomplete inhibition of FLT3 or the emergence of resistant mutations treated with these older agents. We previously developed a series of 3-phenyl-1H-5-pyrazolylamine derivatives as highly potent and selective FLT3 inhibitors with good in vivo efficacy using an intravenous (IV) route. However, the poor bioavailability of these pyrazole compounds limits the development of these promising antileukemic compounds for clinical use. Herein, we describe a novel class of 5-phenyl-thiazol-2-ylamine compounds that are multi-targeted FLT3 inhibitors. From this class of compounds, compound 7h was very potent against AML cell lines and exhibited excellent oral efficacy in AML xenograft models. In addition, further studies demonstrated that compound 7h exhibited potent in vitro and in vivo activities against clinically relevant AC220 (3)-resistant kinase domain mutants of FLT3-ITD. (C) 2015 Published by Elsevier Masson SAS.DOI:10.1016/j.ejmech.2015.05.008
-
作为产物:描述:参考文献:名称:[5,5] Sigmatropic shift of N-phenyl-N′-(2-thiazolyl)hydrazines and N,N′-bis(2-thiazolyl)hydrazines into 2-amino-5-(p-aminophenyl)thiazoles and 5,5′-bis(2-aminothiazole) derivatives摘要:[5,5] Sigmatropic shift of N-phenyl-N'-(2-thiazolyl)hydrazines and N,N'-bis(2-thiazolyl)hydrazines in acid-catalyzed benzidine-type rearrangement into 2-amino-5-(p-aminophenyl)thiazoles and 5,5'-bis(2-aminothiazole) derivatives is described, respectively. (C) 2000 Elsevier Science Ltd. All rights reserved.DOI:10.1016/s0040-4039(00)00493-7
文献信息
-
PYRAZOLE COMPOUNDS AND THIAZOLE COMPOUNDS AS PROTEIN KINASES INHIBITORS申请人:Jiaang Weir-Torn公开号:US20120225880A1公开(公告)日:2012-09-06A compound of formula (I): wherein A, B, D, X, Y, R 1 , R 2 , R 3 , m, p, and q are defined herein. Also disclosed is a method for inhibiting FMS-like tyrosine kinase 3, aurora kinase, or vascular endothelial growth factor receptor.
-
Design, Synthesis, and Anticancer Activity of New Derivatives of Thiazole and Imidazole作者:I. M. El-Deen、E. H. Elsayed、M. El-Ahwany、M. S. Abd El-AzezDOI:10.1134/s1070363220120191日期:2020.12Abstract A number of nitrogen heterocyclic derivatives, namely 1,3-thiazole and imidazolidine-2-thione, have been synthesized and their structures confirmed by IR, 1H and 13C NMR, and mass spectra. Cytotoxic activity of some synthesized products has been tested against human hepatocellular carcinoma (HepG2) cell line using the MTT assay. The most potent anti-proliferative agent demonstrates activity
-
Über die Eigenschaften einiger Phenyl-azol-Derivate作者:J. Eckenstein、E. Brogle、E. Sorkin、H. ErlenmeyerDOI:10.1002/hlca.19500330535日期:——Es wird über die Synthesen einiger Phenyl-azol-Derivate berichtet, von denen auf Grund der Struktur eine tuberkulostatische Aktivität zu erwarten war.报道了一些苯基-唑衍生物的合成,基于它们的结构,预期从中可以得到抑菌活性。
-
Design, synthesis and biological evaluation of novel aminothiazoles as antiviral compounds acting against human rhinovirus作者:Anne Décor、Chantal Grand-Maître、Oliver Hucke、Jeff O’Meara、Cyrille Kuhn、Léa Constantineau -Forget、Christian Brochu、Eric Malenfant、Mégan Bertrand-Laperle、Josée Bordeleau、Elise Ghiro、Marc Pesant、Gulrez Fazal、Vida Gorys、Michael Little、Colette Boucher、Sylvain Bordeleau、Pascal Turcotte、Tim Guo、Michel Garneau、Catherine Spickler、Annick GauthierDOI:10.1016/j.bmcl.2013.04.077日期:2013.7We describe here the design, synthesis and biological evaluation of antiviral compounds acting against human rhinovirus (HRV). A series of aminothiazoles demonstrated pan-activity against the HRV genotypes screened and productive structure-activity relationships. A comprehensive investigational library was designed and performed allowing the identification of potent compounds with lower molecular weight and improved ADME profile. 31d-1, 31d-2, 31f showed good exposures in CD-1 mice. The mechanism of action was discovered to be a host target: the lipid kinase phosphatidylinositol 4-kinase III beta (PI4KIII beta). The identification of the pan-HRV active compound 31f combined with a structurally distinct literature compound T-00127-HEV1 allowed the assessment of target related tolerability of inhibiting this kinase for a short period of time in order to prevent HRV replication. (c) 2013 Elsevier Ltd. All rights reserved.
-
Discovery of Conformational Control Inhibitors Switching off the Activated c-KIT and Targeting a Broad Range of Clinically Relevant c-KIT Mutants作者:Tsung-Sheng Wu、Wen-Hsing Lin、Hui-Jen Tsai、Ching-Cheng Hsueh、Tsu Hsu、Pei-Chen Wang、Hui-You Lin、Yi-Hui Peng、Cheng-Tai Lu、Lung-Chun Lee、Chih-Hsiang Tu、Fang-Chun Kung、Hui-Yi Shiao、Teng-Kuang Yeh、Jen-Shin Song、Jia-Yu Chang、Yu-Chieh Su、Li-Tzong Chen、Chiung-Tong Chen、Weir-Torn Jiaang、Su-Ying WuDOI:10.1021/acs.jmedchem.8b01845日期:2019.4.25Drug resistance due to acquired mutations that constitutively activate c-KIT is a significant challenge in the treatment of patients with gastrointestinal stromal tumors (GISTs). Herein, we identified 1-(5-ethyl-isoxazol-3-yl)-3-(4-2-[6-(4-ethylpiperazin-1-yl)pyrimidin-4-ylamino]-thiazol-5-yl}phenyl)urea (10a) as a potent inhibitor against unactivated and activated c-KIT. The binding of 10a induced rearrangements of the DFG motif, alpha C-helix, juxtamembrane domain, and the activation loop to switch the activated c-KIT back to its structurally inactive state. To the best of our knowledge, it is the first structural evidence demonstrating how a compound can inhibit the activated c-KIT by switching back to its inactive state through a sequence of conformational changes. Moreover, 10a can effectively inhibit various c-KIT mutants and the proliferation of several GIST cell lines. The distinct binding features and superior inhibitory potency of 10a, together with its excellent efficacy in the xenograft model, establish 10a as worthy of further clinical evaluation in the advanced GISTs.
表征谱图
-
氢谱1HNMR
-
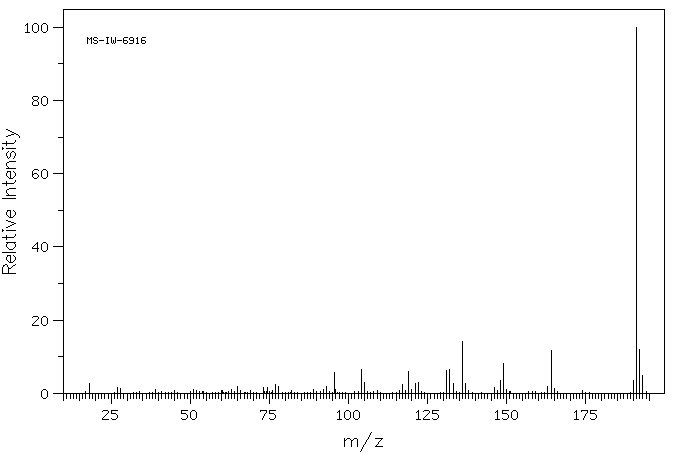
质谱MS
-
碳谱13CNMR
-
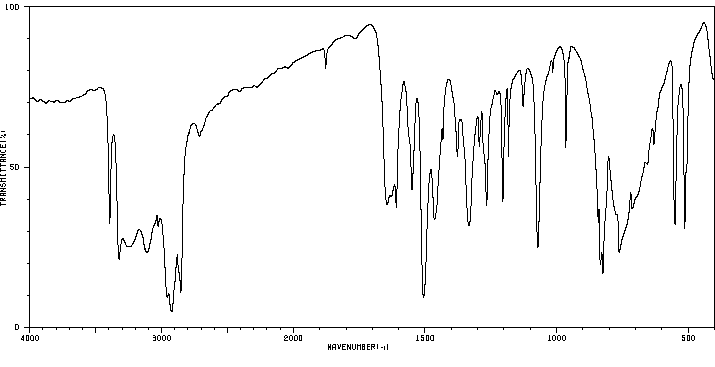
红外IR
-
拉曼Raman
-
峰位数据
-
峰位匹配
-
表征信息
同类化合物
(βS)-β-氨基-4-(4-羟基苯氧基)-3,5-二碘苯甲丙醇
(S,S)-邻甲苯基-DIPAMP
(S)-(-)-7'-〔4(S)-(苄基)恶唑-2-基]-7-二(3,5-二-叔丁基苯基)膦基-2,2',3,3'-四氢-1,1-螺二氢茚
(S)-盐酸沙丁胺醇
(S)-3-(叔丁基)-4-(2,6-二甲氧基苯基)-2,3-二氢苯并[d][1,3]氧磷杂环戊二烯
(S)-2,2'-双[双(3,5-三氟甲基苯基)膦基]-4,4',6,6'-四甲氧基联苯
(S)-1-[3,5-双(三氟甲基)苯基]-3-[1-(二甲基氨基)-3-甲基丁烷-2-基]硫脲
(R)富马酸托特罗定
(R)-(-)-盐酸尼古地平
(R)-(-)-4,12-双(二苯基膦基)[2.2]对环芳烷(1,5环辛二烯)铑(I)四氟硼酸盐
(R)-(+)-7-双(3,5-二叔丁基苯基)膦基7''-[((6-甲基吡啶-2-基甲基)氨基]-2,2'',3,3''-四氢-1,1''-螺双茚满
(R)-(+)-7-双(3,5-二叔丁基苯基)膦基7''-[(4-叔丁基吡啶-2-基甲基)氨基]-2,2'',3,3''-四氢-1,1''-螺双茚满
(R)-(+)-7-双(3,5-二叔丁基苯基)膦基7''-[(3-甲基吡啶-2-基甲基)氨基]-2,2'',3,3''-四氢-1,1''-螺双茚满
(R)-(+)-4,7-双(3,5-二-叔丁基苯基)膦基-7“-[(吡啶-2-基甲基)氨基]-2,2”,3,3'-四氢1,1'-螺二茚满
(R)-3-(叔丁基)-4-(2,6-二苯氧基苯基)-2,3-二氢苯并[d][1,3]氧杂磷杂环戊烯
(R)-2-[((二苯基膦基)甲基]吡咯烷
(R)-1-[3,5-双(三氟甲基)苯基]-3-[1-(二甲基氨基)-3-甲基丁烷-2-基]硫脲
(N-(4-甲氧基苯基)-N-甲基-3-(1-哌啶基)丙-2-烯酰胺)
(5-溴-2-羟基苯基)-4-氯苯甲酮
(5-溴-2-氯苯基)(4-羟基苯基)甲酮
(5-氧代-3-苯基-2,5-二氢-1,2,3,4-oxatriazol-3-鎓)
(4S,5R)-4-甲基-5-苯基-1,2,3-氧代噻唑烷-2,2-二氧化物-3-羧酸叔丁酯
(4S,4''S)-2,2''-亚环戊基双[4,5-二氢-4-(苯甲基)恶唑]
(4-溴苯基)-[2-氟-4-[6-[甲基(丙-2-烯基)氨基]己氧基]苯基]甲酮
(4-丁氧基苯甲基)三苯基溴化磷
(3aR,8aR)-(-)-4,4,8,8-四(3,5-二甲基苯基)四氢-2,2-二甲基-6-苯基-1,3-二氧戊环[4,5-e]二恶唑磷
(3aR,6aS)-5-氧代六氢环戊基[c]吡咯-2(1H)-羧酸酯
(2Z)-3-[[(4-氯苯基)氨基]-2-氰基丙烯酸乙酯
(2S,3S,5S)-5-(叔丁氧基甲酰氨基)-2-(N-5-噻唑基-甲氧羰基)氨基-1,6-二苯基-3-羟基己烷
(2S,2''S,3S,3''S)-3,3''-二叔丁基-4,4''-双(2,6-二甲氧基苯基)-2,2'',3,3''-四氢-2,2''-联苯并[d][1,3]氧杂磷杂戊环
(2S)-(-)-2-{[[[[3,5-双(氟代甲基)苯基]氨基]硫代甲基]氨基}-N-(二苯基甲基)-N,3,3-三甲基丁酰胺
(2S)-2-[[[[[((1S,2S)-2-氨基环己基]氨基]硫代甲基]氨基]-N-(二苯甲基)-N,3,3-三甲基丁酰胺
(2S)-2-[[[[[[((1R,2R)-2-氨基环己基]氨基]硫代甲基]氨基]-N-(二苯甲基)-N,3,3-三甲基丁酰胺
(2-硝基苯基)磷酸三酰胺
(2,6-二氯苯基)乙酰氯
(2,3-二甲氧基-5-甲基苯基)硼酸
(1S,2S,3S,5S)-5-叠氮基-3-(苯基甲氧基)-2-[(苯基甲氧基)甲基]环戊醇
(1S,2S,3R,5R)-2-(苄氧基)甲基-6-氧杂双环[3.1.0]己-3-醇
(1-(4-氟苯基)环丙基)甲胺盐酸盐
(1-(3-溴苯基)环丁基)甲胺盐酸盐
(1-(2-氯苯基)环丁基)甲胺盐酸盐
(1-(2-氟苯基)环丙基)甲胺盐酸盐
(1-(2,6-二氟苯基)环丙基)甲胺盐酸盐
(-)-去甲基西布曲明
龙蒿油
龙胆酸钠
龙胆酸叔丁酯
龙胆酸
龙胆紫-d6
龙胆紫

联系我们



关注我们

公众号

