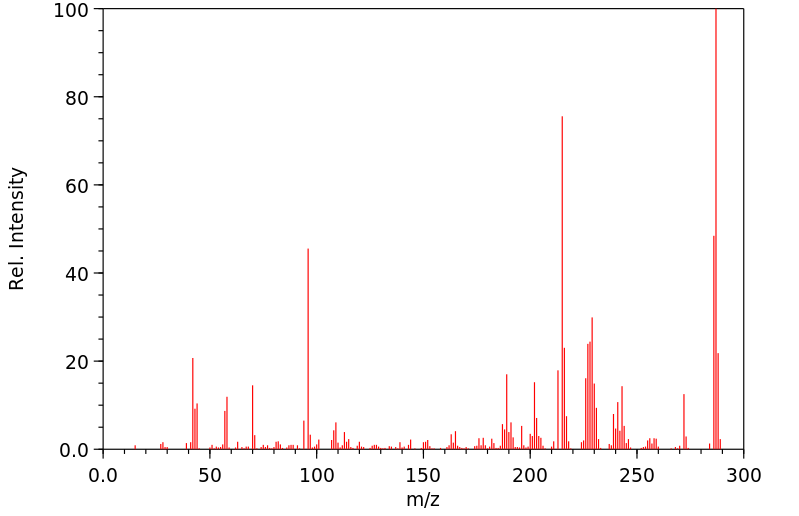
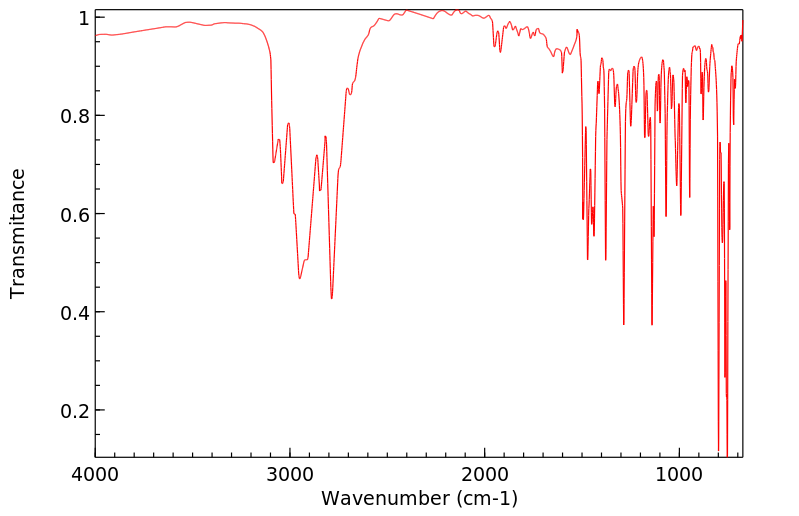
In humans, the metabolism of a number of tertiary amine containing pharmacological agents to quaternary ammonium linked glucuronides, catalyzed by UDP-glucuronosyltransferase (UGT), represents a unique and important metabolic pathway for these compounds. A full length cDNA encoding human UGTl.4 (the so-called minor human bilirubin UGT) was inserted into the expression vector pREP9 and transfected into human embryonic kidney 293 cells, and stable transfectants were obtained after geneticin selection. As expected, the expressed protein had low catalytic activity toward bilirubin. However, expressed human UGTl.4 protein exhibited glucuronidation activity toward tertiary amine substrates, such as imipramine, cyproheptadine, tripelennamine, and chlorpromazine, which form quaternary ammonium linked glucuronides. Carcinogenic primary amines (beta-naphthylamine, benzidine, and 4-aminobiphenyl) also reacted with the expressed UGTl.4 protein at rates approximately 10-fold higher than the rates for quaternary ammonium glucuronide formation. Although a number of other UGT gene products are capable of catalyzing the glucuronidation of primary amine substrates, expressed human UGTl.4 protein is the only UGT isoform that has been shown to conjugate tertiary amine substrates, forming quaternary ammonium linked glucuronides.
Hepatic (cytochrome P-450 system) and some renal.
Route of Elimination: After a single 4 mg oral dose of14C-labelled cyproheptadine HCl in normal subjects, given as tablets 2% to 20% of the radioactivity was excreted in the stools. At least 40% of the administered radioactivity was excreted in the urine.
Cyproheptadine competes with free histamine for binding at HA-receptor sites. This antagonizes the effects of histamine on HA-receptors, leading to a reduction of the negative symptoms brought on by histamine HA-receptor binding. Cyproheptadine also competes with serotonin at receptor sites in smooth muscle in the intestines and other locations. Antagonism of serotonin on the appetite center of the hypothalamus may account for Cyproheptadine's ability to stimulate appetite.
Unlike most first generation antihistamines, cyproheptadine has been associated with several instances of clinically apparent liver injury. The few cases that have been described had a time to onset of 1 to 6 weeks and a cholestatic or mixed pattern of liver enzyme elevations. Immunoallergic and autoimmune features were not present and most patients recovered rapidly without residual. Acute liver failure due to cyproheptadine has not been described.
A single study examining the difference in absorption of orally administered versus sublingually administered cyproheptadine in five healthy males demonstrated a mean Cmax of 30.0 mcg/L and 4.0 mcg/L, respectively, and a mean AUC of 209 mcg.h/L and 25 mcg.h/L, respectively. The Tmax of orally and sublingually administered cyproheptadine was 4 hours and 9.6 hours, respectively.
Approximately 2-20% of the radioactivity from an orally administered radio-labeled dose of cyproheptadine is excreted in the feces, of which approximately 34% is unchanged parent drug (less than 5.7% of the total dose). At least 40% of radioactivity is recovered in the urine.
H1 antagonists are eliminated more rapidly by children than by adults and more slowly in those with severe liver disease. /H1 Receptor Antagonists/
来源:Hazardous Substances Data Bank (HSDB)
吸收、分配和排泄
H1受体拮抗剂从胃肠道吸收良好。口服给药后,2到3小时内达到血浆峰值浓度...。/H1受体拮抗剂/
The H1 antagonists are well absorbed from the gastrointestinal tract. Following oral administration, peak plasma concentrations are achieved in 2 to 3 hours ... . /H1 Receptor Antagonists/
To investigate the pharmacokinetics of cyproheptadine (CPH) and its metabolites, the plasma concn and urinary excretion of CPH and its detectable metabolites were determined after iv admin of parent or synthesized metabolites to rats. The plasma CPH concn time course was subjected to biexponential calculation following the iv admin of CPH, producing the temporal and low plasma concn of desmethylcyproheptadine (DMCPH) and the sustained plasma concn of desmethylcyproheptadine epoxide (DMCPHepo). DMCPH was also eliminated according to the biexponential equation, after iv admin of performed DMCPH, forming DMCPHepo in plasma. On the other hand, no detectable DMCPHepo was found in plasma after the iv admin of cyproheptadine epoxide (CPHepo). All cmpd administered had large distribution volumes and were most entirely excreted as DMCPHepo in urine; this excretion continued for a long time. However, the urinary excretion pattern of DMCPHepo after CPHepo was different from those after CPH and MCPH. The mean residence times of the epoxidized metabolites estimated from the urinary data were much longer than those from the plasma concn data, suggesting either a gradual reflux of the metabolites from a tissue depot into systemic circulation under those plasma concn close of detection limit, or one interaction which delays excretion into the urine. This study suggests that both metabolic pathways of CPH, through DMCPH nd CPHepo, to DMCPHepo are possible, but that the demethylation largely occurs prior to epoxidation; also that the extensive and persistent distribution of DMCPHepo to tissues may relate to the toxicity of CPH reported in rats.
3-Aminocyclopentanecarboxamides as modulators of chemokine receptors
申请人:Xue Chu-Biao
公开号:US20060004018A1
公开(公告)日:2006-01-05
The present invention is directed to compounds of Formula I:
which are modulators of chemokine receptors. The compounds of the invention, and compositions thereof, are useful in the treatment of diseases related to chemokine receptor expression and/or activity.
Benzoxazinyl-amidocyclopentyl-heterocyclic modulators of chemokine receptors
申请人:Goble D. Stephen
公开号:US20070238723A1
公开(公告)日:2007-10-11
Cyclopentyl compounds linked to a benzoxazinyl group through an amido moiety utilizing the ring nitrogen of the benzoxazine, and further substituted with a heterocyclic moiety, such compounds represented by formula I:
which are used to modulate the CCR-2 chemokine receptor to prevent or treat inflammatory and immunoregulatory disorders and diseases, allergic diseases, atopic conditions including allergic rhinitis, dermatitis, conjunctivitis, and asthma, as well as autoimmune pathologies such as rheumatoid arthritis and atherosclerosis; and pharmaceutical compositions comprising these compounds and the use of these compounds and compositions.
[EN] AMINE-LINKED C3-GLUTARIMIDE DEGRONIMERS FOR TARGET PROTEIN DEGRADATION<br/>[FR] DÉGRONIMÈRES DE C3-GLUTARIMIDE LIÉS À UNE AMINE POUR LA DÉGRADATION DE PROTÉINES CIBLES
申请人:C4 THERAPEUTICS INC
公开号:WO2017197051A1
公开(公告)日:2017-11-16
This invention provides amine-linked C3-glutarimide Degronimers and Degrons for therapeutic applications as described further herein, and methods of use and compositions thereof as well as methods for their preparation.
Imidazole and benzimidazole derivatives useful as histamine H3 antagonists
申请人:Aslanian G. Robert
公开号:US20060166960A1
公开(公告)日:2006-07-27
Disclosed are compounds of the formula
or a pharmaceutically acceptable salt or solvate thereof, wherein: n is 2-5;
R is R
3
-aryl, R
3
-heteroaryl, R
3
-cycloalkyl, R
3
-heterocycloalkyl, alkyl, haloalkyl, —OR
4
, —SR
4
or —S(O)
1-2
R
5
;
R
1
and R
2
are H or optionally substituted phenyl or optionally substituted
and X is —O— or —S—;
or R
1
and R
2
, together with the carbon atoms to which they are attached form optionally substituted
and X is —O—, —S— or —NR
7
—;
Z is
and the remaining variables are as defined in the specification; also disclosed are pharmaceutical compositions comprising the compounds of formula I; also disclosed are methods of treating allergy, allergy-induced airway responses, congestion, obesity and metabolic syndrome using the compounds of Formula I, as well as combinations with other drugs useful for treating those diseases.
[EN] 1-(4-PIPERIDINYL) BENZIMIDAZOLONES AS HISTAMINE H3 ANTAGONISTS<br/>[FR] 1-(4-PIPERIDINYL) BENZIMIDAZOLONES UTILISES EN TANT QU'ANTAGONISTES DU RECEPTEUR H3 DE L'HISTAMINE
申请人:SCHERING CORP
公开号:WO2003103669A1
公开(公告)日:2003-12-18
Disclosed are histamine H3 antagonists of the formula (I) wherein
R1 is benzimidazolone derivative, M1 and M2 are
optionally substituted carbon or nitrogen, R2 includes optionally
substituted aryl or heteroaryl, and the remaining variables are as defined in
the specification. Also disclosed are pharmaceutical compositions comprising
the compounds of formula (I). Also disclosed are methods of treating various
diseases or conditions, such as, for example, allergy, allergy-induced airway
responses, and congestion (e.g., nasal congestion) using the compounds of Formula
(I). Also disclosed are methods of treating various diseases or conditions,
such as, for example, allergy, allergy-induced airway responses, and congestion
(e.g., nasal congestion) using the compounds of formula (I) in combination with
a H1 receptor antagonist.