
2-[(4-二甲基氨基苯基)亚甲基氨基]苯酚 | 3230-43-1
中文名称
2-[(4-二甲基氨基苯基)亚甲基氨基]苯酚
中文别名
——
英文名称
2-(4-dimethylamino-benzylidenamino)-phenol
英文别名
2-(4-(dimethylamino)benzylideneamino)phenol;2-Hydroxy-N-<4-dimethylamino-benzyliden>-anilin;2-<4-Dimethylamino-benzylidenamino>-phenol;N-<4-Dimethylamino-benzyliden>-2-hydroxy-anilin;o-(p-(Dimethylamino)benzylideneamino)phenol;2-[[4-(dimethylamino)phenyl]methylideneamino]phenol
![2-[(4-二甲基氨基苯基)亚甲基氨基]苯酚化学式](data:image/svg+xml;base64,PD94bWwgdmVyc2lvbj0iMS4wIiBlbmNvZGluZz0iVVRGLTgiPz4KPHN2ZyB4bWxucz0iaHR0cDovL3d3dy53My5vcmcvMjAwMC9zdmciIHhtbG5zOnhsaW5rPSJodHRwOi8vd3d3LnczLm9yZy8xOTk5L3hsaW5rIiB3aWR0aD0iMzAwIiBoZWlnaHQ9IjMwMCIgdmlld0JveD0iMCAwIDMwMCAzMDAiPgo8ZGVmcz4KPGc+CjxnIGlkPSJnbHlwaC0wLTAiPgo8cGF0aCBkPSJNIDAuNjg3NSAyLjQzNzUgTCAwLjY4NzUgLTkuNzAzMTI1IEwgNy41NjI1IC05LjcwMzEyNSBMIDcuNTYyNSAyLjQzNzUgWiBNIDEuNDUzMTI1IDEuNjcxODc1IEwgNi43OTY4NzUgMS42NzE4NzUgTCA2Ljc5Njg3NSAtOC45Mzc1IEwgMS40NTMxMjUgLTguOTM3NSBaIE0gMS40NTMxMjUgMS42NzE4NzUgIi8+CjwvZz4KPGcgaWQ9ImdseXBoLTAtMSI+CjxwYXRoIGQ9Ik0gMS4zNDM3NSAtMTAuMDMxMjUgTCAzLjE3MTg3NSAtMTAuMDMxMjUgTCA3LjYyNSAtMS42NDA2MjUgTCA3LjYyNSAtMTAuMDMxMjUgTCA4Ljk1MzEyNSAtMTAuMDMxMjUgTCA4Ljk1MzEyNSAwIEwgNy4xMjUgMCBMIDIuNjcxODc1IC04LjM5MDYyNSBMIDIuNjcxODc1IDAgTCAxLjM0Mzc1IDAgWiBNIDEuMzQzNzUgLTEwLjAzMTI1ICIvPgo8L2c+CjxnIGlkPSJnbHlwaC0wLTIiPgo8cGF0aCBkPSJNIDUuNDIxODc1IC05LjEwOTM3NSBDIDQuNDQxNDA2IC05LjEwOTM3NSAzLjY2MDE1NiAtOC43MzgyODEgMy4wNzgxMjUgLTggQyAyLjQ5MjE4OCAtNy4yNjk1MzEgMi4yMDMxMjUgLTYuMjY5NTMxIDIuMjAzMTI1IC01IEMgMi4yMDMxMjUgLTMuNzM4MjgxIDIuNDkyMTg4IC0yLjczODI4MSAzLjA3ODEyNSAtMiBDIDMuNjYwMTU2IC0xLjI2OTUzMSA0LjQ0MTQwNiAtMC45MDYyNSA1LjQyMTg3NSAtMC45MDYyNSBDIDYuNDEwMTU2IC0wLjkwNjI1IDcuMTkxNDA2IC0xLjI2OTUzMSA3Ljc2NTYyNSAtMiBDIDguMzM1OTM4IC0yLjczODI4MSA4LjYyNSAtMy43MzgyODEgOC42MjUgLTUgQyA4LjYyNSAtNi4yNjk1MzEgOC4zMzU5MzggLTcuMjY5NTMxIDcuNzY1NjI1IC04IEMgNy4xOTE0MDYgLTguNzM4MjgxIDYuNDEwMTU2IC05LjEwOTM3NSA1LjQyMTg3NSAtOS4xMDkzNzUgWiBNIDUuNDIxODc1IC0xMC4yMTg3NSBDIDYuODI4MTI1IC0xMC4yMTg3NSA3Ljk1MzEyNSAtOS43NDIxODggOC43OTY4NzUgLTguNzk2ODc1IEMgOS42NDA2MjUgLTcuODU5Mzc1IDEwLjA2MjUgLTYuNTkzNzUgMTAuMDYyNSAtNSBDIDEwLjA2MjUgLTMuNDI1NzgxIDkuNjQwNjI1IC0yLjE2NDA2MiA4Ljc5Njg3NSAtMS4yMTg3NSBDIDcuOTUzMTI1IC0wLjI4MTI1IDYuODI4MTI1IDAuMTg3NSA1LjQyMTg3NSAwLjE4NzUgQyA0LjAxNTYyNSAwLjE4NzUgMi44ODI4MTIgLTAuMjgxMjUgMi4wMzEyNSAtMS4yMTg3NSBDIDEuMTg3NSAtMi4xNTYyNSAwLjc2NTYyNSAtMy40MTQwNjIgMC43NjU2MjUgLTUgQyAwLjc2NTYyNSAtNi41OTM3NSAxLjE4NzUgLTcuODU5Mzc1IDIuMDMxMjUgLTguNzk2ODc1IEMgMi44ODI4MTIgLTkuNzQyMTg4IDQuMDE1NjI1IC0xMC4yMTg3NSA1LjQyMTg3NSAtMTAuMjE4NzUgWiBNIDUuNDIxODc1IC0xMC4yMTg3NSAiLz4KPC9nPgo8ZyBpZD0iZ2x5cGgtMC0zIj4KPHBhdGggZD0iTSAxLjM0Mzc1IC0xMC4wMzEyNSBMIDIuNzAzMTI1IC0xMC4wMzEyNSBMIDIuNzAzMTI1IC01LjkyMTg3NSBMIDcuNjQwNjI1IC01LjkyMTg3NSBMIDcuNjQwNjI1IC0xMC4wMzEyNSBMIDkgLTEwLjAzMTI1IEwgOSAwIEwgNy42NDA2MjUgMCBMIDcuNjQwNjI1IC00Ljc4MTI1IEwgMi43MDMxMjUgLTQuNzgxMjUgTCAyLjcwMzEyNSAwIEwgMS4zNDM3NSAwIFogTSAxLjM0Mzc1IC0xMC4wMzEyNSAiLz4KPC9nPgo8ZyBpZD0iZ2x5cGgtMC00Ij4KPHBhdGggZD0iTSA4Ljg1OTM3NSAtOS4yNjU2MjUgTCA4Ljg1OTM3NSAtNy44MjgxMjUgQyA4LjQxMDE1NiAtOC4yNTM5MDYgNy45MjU3ODEgLTguNTcwMzEyIDcuNDA2MjUgLTguNzgxMjUgQyA2Ljg4MjgxMiAtOC45ODgyODEgNi4zMzIwMzEgLTkuMDkzNzUgNS43NSAtOS4wOTM3NSBDIDQuNjAxNTYyIC05LjA5Mzc1IDMuNzIyNjU2IC04Ljc0MjE4OCAzLjEwOTM3NSAtOC4wNDY4NzUgQyAyLjUwMzkwNiAtNy4zNDc2NTYgMi4yMDMxMjUgLTYuMzMyMDMxIDIuMjAzMTI1IC01IEMgMi4yMDMxMjUgLTMuNjg3NSAyLjUwMzkwNiAtMi42NzU3ODEgMy4xMDkzNzUgLTEuOTY4NzUgQyAzLjcyMjY1NiAtMS4yNjk1MzEgNC42MDE1NjIgLTAuOTIxODc1IDUuNzUgLTAuOTIxODc1IEMgNi4zMzIwMzEgLTAuOTIxODc1IDYuODgyODEyIC0xLjAyMzQzOCA3LjQwNjI1IC0xLjIzNDM3NSBDIDcuOTI1NzgxIC0xLjQ0MTQwNiA4LjQxMDE1NiAtMS43NTc4MTIgOC44NTkzNzUgLTIuMTg3NSBMIDguODU5Mzc1IC0wLjc2NTYyNSBDIDguMzkwNjI1IC0wLjQ0MTQwNiA3Ljg5MDYyNSAtMC4yMDMxMjUgNy4zNTkzNzUgLTAuMDQ2ODc1IEMgNi44MjgxMjUgMC4xMDkzNzUgNi4yNjU2MjUgMC4xODc1IDUuNjcxODc1IDAuMTg3NSBDIDQuMTQ4NDM4IDAuMTg3NSAyLjk1MzEyNSAtMC4yNzM0MzggMi4wNzgxMjUgLTEuMjAzMTI1IEMgMS4yMDMxMjUgLTIuMTI4OTA2IDAuNzY1NjI1IC0zLjM5NDUzMSAwLjc2NTYyNSAtNSBDIDAuNzY1NjI1IC02LjYxMzI4MSAxLjIwMzEyNSAtNy44ODI4MTIgMi4wNzgxMjUgLTguODEyNSBDIDIuOTUzMTI1IC05Ljc1IDQuMTQ4NDM4IC0xMC4yMTg3NSA1LjY3MTg3NSAtMTAuMjE4NzUgQyA2LjI3MzQzOCAtMTAuMjE4NzUgNi44NDM3NSAtMTAuMTMyODEyIDcuMzc1IC05Ljk2ODc1IEMgNy45MDYyNSAtOS44MTI1IDguMzk4NDM4IC05LjU3ODEyNSA4Ljg1OTM3NSAtOS4yNjU2MjUgWiBNIDguODU5Mzc1IC05LjI2NTYyNSAiLz4KPC9nPgo8ZyBpZD0iZ2x5cGgtMS0wIj4KPHBhdGggZD0iTSAwLjQ1MzEyNSAxLjYyNSBMIDAuNDUzMTI1IC02LjQ4NDM3NSBMIDUuMDQ2ODc1IC02LjQ4NDM3NSBMIDUuMDQ2ODc1IDEuNjI1IFogTSAwLjk2ODc1IDEuMTA5Mzc1IEwgNC41NDY4NzUgMS4xMDkzNzUgTCA0LjU0Njg3NSAtNS45Njg3NSBMIDAuOTY4NzUgLTUuOTY4NzUgWiBNIDAuOTY4NzUgMS4xMDkzNzUgIi8+CjwvZz4KPGcgaWQ9ImdseXBoLTEtMSI+CjxwYXRoIGQ9Ik0gMy43MzQzNzUgLTMuNjA5Mzc1IEMgNC4xNjAxNTYgLTMuNTE1NjI1IDQuNDkyMTg4IC0zLjMyMDMxMiA0LjczNDM3NSAtMy4wMzEyNSBDIDQuOTg0Mzc1IC0yLjczODI4MSA1LjEwOTM3NSAtMi4zNzg5MDYgNS4xMDkzNzUgLTEuOTUzMTI1IEMgNS4xMDkzNzUgLTEuMjg1MTU2IDQuODc4OTA2IC0wLjc2OTUzMSA0LjQyMTg3NSAtMC40MDYyNSBDIDMuOTcyNjU2IC0wLjA1MDc4MTIgMy4zMjgxMjUgMC4xMjUgMi40ODQzNzUgMC4xMjUgQyAyLjIwMzEyNSAwLjEyNSAxLjkxMDE1NiAwLjA5NzY1NjIgMS42MDkzNzUgMC4wNDY4NzUgQyAxLjMxNjQwNiAtMC4wMDM5MDYyNSAxLjAxNTYyNSAtMC4wODU5Mzc1IDAuNzAzMTI1IC0wLjIwMzEyNSBMIDAuNzAzMTI1IC0xLjA3ODEyNSBDIDAuOTUzMTI1IC0wLjkyOTY4OCAxLjIyMjY1NiAtMC44MTY0MDYgMS41MTU2MjUgLTAuNzM0Mzc1IEMgMS44MTY0MDYgLTAuNjYwMTU2IDIuMTMyODEyIC0wLjYyNSAyLjQ2ODc1IC0wLjYyNSBDIDMuMDMxMjUgLTAuNjI1IDMuNDU3MDMxIC0wLjczODI4MSAzLjc1IC0wLjk2ODc1IEMgNC4wNTA3ODEgLTEuMTk1MzEyIDQuMjAzMTI1IC0xLjUyMzQzOCA0LjIwMzEyNSAtMS45NTMxMjUgQyA0LjIwMzEyNSAtMi4zNDc2NTYgNC4wNjI1IC0yLjY1NjI1IDMuNzgxMjUgLTIuODc1IEMgMy41MDc4MTIgLTMuMDkzNzUgMy4xMjg5MDYgLTMuMjAzMTI1IDIuNjQwNjI1IC0zLjIwMzEyNSBMIDEuODU5Mzc1IC0zLjIwMzEyNSBMIDEuODU5Mzc1IC0zLjk1MzEyNSBMIDIuNjcxODc1IC0zLjk1MzEyNSBDIDMuMTE3MTg4IC0zLjk1MzEyNSAzLjQ1NzAzMSAtNC4wMzkwNjIgMy42ODc1IC00LjIxODc1IEMgMy45MjU3ODEgLTQuMzk0NTMxIDQuMDQ2ODc1IC00LjY0ODQzOCA0LjA0Njg3NSAtNC45ODQzNzUgQyA0LjA0Njg3NSAtNS4zMjgxMjUgMy45MjE4NzUgLTUuNTkzNzUgMy42NzE4NzUgLTUuNzgxMjUgQyAzLjQyOTY4OCAtNS45Njg3NSAzLjA4NTkzOCAtNi4wNjI1IDIuNjQwNjI1IC02LjA2MjUgQyAyLjM5MDYyNSAtNi4wNjI1IDIuMTE3MTg4IC02LjAzMTI1IDEuODI4MTI1IC01Ljk2ODc1IEMgMS41NDY4NzUgLTUuOTE0MDYyIDEuMjM4MjgxIC01LjgzMjAzMSAwLjkwNjI1IC01LjcxODc1IEwgMC45MDYyNSAtNi41MzEyNSBDIDEuMjUgLTYuNjI1IDEuNTY2NDA2IC02LjY5MTQwNiAxLjg1OTM3NSAtNi43MzQzNzUgQyAyLjE2MDE1NiAtNi43ODUxNTYgMi40NDUzMTIgLTYuODEyNSAyLjcxODc1IC02LjgxMjUgQyAzLjQwNjI1IC02LjgxMjUgMy45NDUzMTIgLTYuNjU2MjUgNC4zNDM3NSAtNi4zNDM3NSBDIDQuNzUgLTYuMDMxMjUgNC45NTMxMjUgLTUuNjA5Mzc1IDQuOTUzMTI1IC01LjA3ODEyNSBDIDQuOTUzMTI1IC00LjcxMDkzOCA0Ljg0Mzc1IC00LjM5ODQzOCA0LjYyNSAtNC4xNDA2MjUgQyA0LjQxNDA2MiAtMy44OTA2MjUgNC4xMTcxODggLTMuNzEwOTM4IDMuNzM0Mzc1IC0zLjYwOTM3NSBaIE0gMy43MzQzNzUgLTMuNjA5Mzc1ICIvPgo8L2c+CjwvZz4KPC9kZWZzPgo8cGF0aCBmaWxsPSJub25lIiBzdHJva2Utd2lkdGg9IjAuMDMzMzMzMyIgc3Ryb2tlLWxpbmVjYXA9ImJ1dHQiIHN0cm9rZS1saW5lam9pbj0ibWl0ZXIiIHN0cm9rZT0icmdiKDAlLCAwJSwgMCUpIiBzdHJva2Utb3BhY2l0eT0iMSIgc3Ryb2tlLW1pdGVybGltaXQ9IjEwIiBkPSJNIDUuOTk1MjkzIDEuNzQwMTg0IEwgNi41MDQ5MzQgMC44NTc2MTYgIiB0cmFuc2Zvcm09Im1hdHJpeCgzNC40Mjk4NjcsIDAsIDAsIDM0LjQyOTg2NywgMjEuODk1MzUxLCA5MC4wOTc0MDkpIi8+CjxwYXRoIGZpbGw9Im5vbmUiIHN0cm9rZS13aWR0aD0iMC4wMzMzMzMzIiBzdHJva2UtbGluZWNhcD0iYnV0dCIgc3Ryb2tlLWxpbmVqb2luPSJtaXRlciIgc3Ryb2tlPSJyZ2IoMCUsIDAlLCAwJSkiIHN0cm9rZS1vcGFjaXR5PSIxIiBzdHJva2UtbWl0ZXJsaW1pdD0iMTAiIGQ9Ik0gNi4wMDk3MDIgMS43MzE5MDIgTCA1LjIyMDUwNyAxLjczMTkwMiAiIHRyYW5zZm9ybT0ibWF0cml4KDM0LjQyOTg2NywgMCwgMCwgMzQuNDI5ODY3LCAyMS44OTUzNTEsIDkwLjA5NzQwOSkiLz4KPHBhdGggZmlsbD0ibm9uZSIgc3Ryb2tlLXdpZHRoPSIwLjAzMzMzMzMiIHN0cm9rZS1saW5lY2FwPSJidXR0IiBzdHJva2UtbGluZWpvaW49Im1pdGVyIiBzdHJva2U9InJnYigwJSwgMCUsIDAlKSIgc3Ryb2tlLW9wYWNpdHk9IjEiIHN0cm9rZS1taXRlcmxpbWl0PSIxMCIgZD0iTSA2LjAwMDA1OCAxLjczMTkwMiBMIDYuNTA0OTM0IDIuNjA2MTg4ICIgdHJhbnNmb3JtPSJtYXRyaXgoMzQuNDI5ODY3LCAwLCAwLCAzNC40Mjk4NjcsIDIxLjg5NTM1MSwgOTAuMDk3NDA5KSIvPgo8cGF0aCBmaWxsPSJub25lIiBzdHJva2Utd2lkdGg9IjAuMDMzMzMzMyIgc3Ryb2tlLWxpbmVjYXA9ImJ1dHQiIHN0cm9rZS1saW5lam9pbj0ibWl0ZXIiIHN0cm9rZT0icmdiKDAlLCAwJSwgMCUpIiBzdHJva2Utb3BhY2l0eT0iMSIgc3Ryb2tlLW1pdGVybGltaXQ9IjEwIiBkPSJNIDYuMTkyNTkyIDEuNzMxOTAyIEwgNi42MDExNDQgMi40Mzk1MjMgIiB0cmFuc2Zvcm09Im1hdHJpeCgzNC40Mjk4NjcsIDAsIDAsIDM0LjQyOTg2NywgMjEuODk1MzUxLCA5MC4wOTc0MDkpIi8+CjxwYXRoIGZpbGw9Im5vbmUiIHN0cm9rZS13aWR0aD0iMC4wMzMzMzMzIiBzdHJva2UtbGluZWNhcD0iYnV0dCIgc3Ryb2tlLWxpbmVqb2luPSJtaXRlciIgc3Ryb2tlPSJyZ2IoMCUsIDAlLCAwJSkiIHN0cm9rZS1vcGFjaXR5PSIxIiBzdHJva2UtbWl0ZXJsaW1pdD0iMTAiIGQ9Ik0gNi41MDAwNTYgMC44NjU4OTggTCA3LjUwOTY5NCAwLjg2NTg5OCAiIHRyYW5zZm9ybT0ibWF0cml4KDM0LjQyOTg2NywgMCwgMCwgMzQuNDI5ODY3LCAyMS44OTUzNTEsIDkwLjA5NzQwOSkiLz4KPHBhdGggZmlsbD0ibm9uZSIgc3Ryb2tlLXdpZHRoPSIwLjAzMzMzMzMiIHN0cm9rZS1saW5lY2FwPSJidXR0IiBzdHJva2UtbGluZWpvaW49Im1pdGVyIiBzdHJva2U9InJnYigwJSwgMCUsIDAlKSIgc3Ryb2tlLW9wYWNpdHk9IjEiIHN0cm9rZS1taXRlcmxpbWl0PSIxMCIgZD0iTSA2LjU5NjI2NiAxLjAzMjU2NCBMIDcuNDEzNDg0IDEuMDMyNTY0ICIgdHJhbnNmb3JtPSJtYXRyaXgoMzQuNDI5ODY3LCAwLCAwLCAzNC40Mjk4NjcsIDIxLjg5NTM1MSwgOTAuMDk3NDA5KSIvPgo8cGF0aCBmaWxsPSJub25lIiBzdHJva2Utd2lkdGg9IjAuMDMzMzMzMyIgc3Ryb2tlLWxpbmVjYXA9ImJ1dHQiIHN0cm9rZS1saW5lam9pbj0ibWl0ZXIiIHN0cm9rZT0icmdiKDAlLCAwJSwgMCUpIiBzdHJva2Utb3BhY2l0eT0iMSIgc3Ryb2tlLW1pdGVybGltaXQ9IjEwIiBkPSJNIDYuNTA0OTM0IDAuODc0Mjk0IEwgNi4xNjIyOTkgMC4yODA5MjIgIiB0cmFuc2Zvcm09Im1hdHJpeCgzNC40Mjk4NjcsIDAsIDAsIDM0LjQyOTg2NywgMjEuODk1MzUxLCA5MC4wOTc0MDkpIi8+CjxwYXRoIGZpbGw9Im5vbmUiIHN0cm9rZS13aWR0aD0iMC4wMzMzMzMzIiBzdHJva2UtbGluZWNhcD0iYnV0dCIgc3Ryb2tlLWxpbmVqb2luPSJtaXRlciIgc3Ryb2tlPSJyZ2IoMCUsIDAlLCAwJSkiIHN0cm9rZS1vcGFjaXR5PSIxIiBzdHJva2UtbWl0ZXJsaW1pdD0iMTAiIGQ9Ik0gNC43NzQyODggMS45NTYzMTYgTCA0LjU2NzQ1OSAyLjY0Nzk0ICIgdHJhbnNmb3JtPSJtYXRyaXgoMzQuNDI5ODY3LCAwLCAwLCAzNC40Mjk4NjcsIDIxLjg5NTM1MSwgOTAuMDk3NDA5KSIvPgo8cGF0aCBmaWxsPSJub25lIiBzdHJva2Utd2lkdGg9IjAuMDMzMzMzMyIgc3Ryb2tlLWxpbmVjYXA9ImJ1dHQiIHN0cm9rZS1saW5lam9pbj0ibWl0ZXIiIHN0cm9rZT0icmdiKDAlLCAwJSwgMCUpIiBzdHJva2Utb3BhY2l0eT0iMSIgc3Ryb2tlLW1pdGVybGltaXQ9IjEwIiBkPSJNIDQuOTE4NjAzIDIuMDM5NzA2IEwgNC40MjMxNDQgMi41NjQ1NSAiIHRyYW5zZm9ybT0ibWF0cml4KDM0LjQyOTg2NywgMCwgMCwgMzQuNDI5ODY3LCAyMS44OTUzNTEsIDkwLjA5NzQwOSkiLz4KPHBhdGggZmlsbD0ibm9uZSIgc3Ryb2tlLXdpZHRoPSIwLjAzMzMzMzMiIHN0cm9rZS1saW5lY2FwPSJidXR0IiBzdHJva2UtbGluZWpvaW49Im1pdGVyIiBzdHJva2U9InJnYigwJSwgMCUsIDAlKSIgc3Ryb2tlLW9wYWNpdHk9IjEiIHN0cm9rZS1taXRlcmxpbWl0PSIxMCIgZD0iTSA2LjQ5MDQxMiAyLjU5NzkwNiBMIDcuNTA5Njk0IDIuNTk3OTA2ICIgdHJhbnNmb3JtPSJtYXRyaXgoMzQuNDI5ODY3LCAwLCAwLCAzNC40Mjk4NjcsIDIxLjg5NTM1MSwgOTAuMDk3NDA5KSIvPgo8cGF0aCBmaWxsPSJub25lIiBzdHJva2Utd2lkdGg9IjAuMDMzMzMzMyIgc3Ryb2tlLWxpbmVjYXA9ImJ1dHQiIHN0cm9rZS1saW5lam9pbj0ibWl0ZXIiIHN0cm9rZT0icmdiKDAlLCAwJSwgMCUpIiBzdHJva2Utb3BhY2l0eT0iMSIgc3Ryb2tlLW1pdGVybGltaXQ9IjEwIiBkPSJNIDcuNDk1Mjg1IDAuODU3NjE2IEwgOC4wMDQ5MjcgMS43NDAxODQgIiB0cmFuc2Zvcm09Im1hdHJpeCgzNC40Mjk4NjcsIDAsIDAsIDM0LjQyOTg2NywgMjEuODk1MzUxLCA5MC4wOTc0MDkpIi8+CjxwYXRoIGZpbGw9Im5vbmUiIHN0cm9rZS13aWR0aD0iMC4wMzMzMzMzIiBzdHJva2UtbGluZWNhcD0iYnV0dCIgc3Ryb2tlLWxpbmVqb2luPSJtaXRlciIgc3Ryb2tlPSJyZ2IoMCUsIDAlLCAwJSkiIHN0cm9rZS1vcGFjaXR5PSIxIiBzdHJva2UtbWl0ZXJsaW1pdD0iMTAiIGQ9Ik0gNC41MDk4MjMgMi41OTc5MDYgTCAzLjQ5MDY1NSAyLjU5NzkwNiAiIHRyYW5zZm9ybT0ibWF0cml4KDM0LjQyOTg2NywgMCwgMCwgMzQuNDI5ODY3LCAyMS44OTUzNTEsIDkwLjA5NzQwOSkiLz4KPHBhdGggZmlsbD0ibm9uZSIgc3Ryb2tlLXdpZHRoPSIwLjAzMzMzMzMiIHN0cm9rZS1saW5lY2FwPSJidXR0IiBzdHJva2UtbGluZWpvaW49Im1pdGVyIiBzdHJva2U9InJnYigwJSwgMCUsIDAlKSIgc3Ryb2tlLW9wYWNpdHk9IjEiIHN0cm9rZS1taXRlcmxpbWl0PSIxMCIgZD0iTSA3LjQ5NTI4NSAyLjYwNjE4OCBMIDguMDA0OTI3IDEuNzIzNTA2ICIgdHJhbnNmb3JtPSJtYXRyaXgoMzQuNDI5ODY3LCAwLCAwLCAzNC40Mjk4NjcsIDIxLjg5NTM1MSwgOTAuMDk3NDA5KSIvPgo8cGF0aCBmaWxsPSJub25lIiBzdHJva2Utd2lkdGg9IjAuMDMzMzMzMyIgc3Ryb2tlLWxpbmVjYXA9ImJ1dHQiIHN0cm9rZS1saW5lam9pbj0ibWl0ZXIiIHN0cm9rZT0icmdiKDAlLCAwJSwgMCUpIiBzdHJva2Utb3BhY2l0eT0iMSIgc3Ryb2tlLW1pdGVybGltaXQ9IjEwIiBkPSJNIDcuMzk5MDc1IDIuNDM5NTIzIEwgNy44MTIzOTMgMS43MjM1MDYgIiB0cmFuc2Zvcm09Im1hdHJpeCgzNC40Mjk4NjcsIDAsIDAsIDM0LjQyOTg2NywgMjEuODk1MzUxLCA5MC4wOTc0MDkpIi8+CjxwYXRoIGZpbGw9Im5vbmUiIHN0cm9rZS13aWR0aD0iMC4wMzMzMzMzIiBzdHJva2UtbGluZWNhcD0iYnV0dCIgc3Ryb2tlLWxpbmVqb2luPSJtaXRlciIgc3Ryb2tlPSJyZ2IoMCUsIDAlLCAwJSkiIHN0cm9rZS1vcGFjaXR5PSIxIiBzdHJva2UtbWl0ZXJsaW1pdD0iMTAiIGQ9Ik0gMy41MDAyOTggMi41OTc5MDYgTCAyLjk5NTQyMiAzLjQ3MjE5MiAiIHRyYW5zZm9ybT0ibWF0cml4KDM0LjQyOTg2NywgMCwgMCwgMzQuNDI5ODY3LCAyMS44OTUzNTEsIDkwLjA5NzQwOSkiLz4KPHBhdGggZmlsbD0ibm9uZSIgc3Ryb2tlLXdpZHRoPSIwLjAzMzMzMzMiIHN0cm9rZS1saW5lY2FwPSJidXR0IiBzdHJva2UtbGluZWpvaW49Im1pdGVyIiBzdHJva2U9InJnYigwJSwgMCUsIDAlKSIgc3Ryb2tlLW9wYWNpdHk9IjEiIHN0cm9rZS1taXRlcmxpbWl0PSIxMCIgZD0iTSAzLjMwNzc2NSAyLjU5NzkwNiBMIDIuODk5MjEyIDMuMzA1NTI3ICIgdHJhbnNmb3JtPSJtYXRyaXgoMzQuNDI5ODY3LCAwLCAwLCAzNC40Mjk4NjcsIDIxLjg5NTM1MSwgOTAuMDk3NDA5KSIvPgo8cGF0aCBmaWxsPSJub25lIiBzdHJva2Utd2lkdGg9IjAuMDMzMzMzMyIgc3Ryb2tlLWxpbmVjYXA9ImJ1dHQiIHN0cm9rZS1saW5lam9pbj0ibWl0ZXIiIHN0cm9rZT0icmdiKDAlLCAwJSwgMCUpIiBzdHJva2Utb3BhY2l0eT0iMSIgc3Ryb2tlLW1pdGVybGltaXQ9IjEwIiBkPSJNIDMuNTA1MDYzIDIuNjA2MTg4IEwgMi45OTU0MjIgMS43MjM1MDYgIiB0cmFuc2Zvcm09Im1hdHJpeCgzNC40Mjk4NjcsIDAsIDAsIDM0LjQyOTg2NywgMjEuODk1MzUxLCA5MC4wOTc0MDkpIi8+CjxwYXRoIGZpbGw9Im5vbmUiIHN0cm9rZS13aWR0aD0iMC4wMzMzMzMzIiBzdHJva2UtbGluZWNhcD0iYnV0dCIgc3Ryb2tlLWxpbmVqb2luPSJtaXRlciIgc3Ryb2tlPSJyZ2IoMCUsIDAlLCAwJSkiIHN0cm9rZS1vcGFjaXR5PSIxIiBzdHJva2UtbWl0ZXJsaW1pdD0iMTAiIGQ9Ik0gMy4wMDk5NDUgMy40NjM5MSBMIDEuOTkwMzIyIDMuNDYzOTEgIiB0cmFuc2Zvcm09Im1hdHJpeCgzNC40Mjk4NjcsIDAsIDAsIDM0LjQyOTg2NywgMjEuODk1MzUxLCA5MC4wOTc0MDkpIi8+CjxwYXRoIGZpbGw9Im5vbmUiIHN0cm9rZS13aWR0aD0iMC4wMzMzMzMzIiBzdHJva2UtbGluZWNhcD0iYnV0dCIgc3Ryb2tlLWxpbmVqb2luPSJtaXRlciIgc3Ryb2tlPSJyZ2IoMCUsIDAlLCAwJSkiIHN0cm9rZS1vcGFjaXR5PSIxIiBzdHJva2UtbWl0ZXJsaW1pdD0iMTAiIGQ9Ik0gMy4wMDk5NDUgMS43MzE5MDIgTCAxLjk5MDMyMiAxLjczMTkwMiAiIHRyYW5zZm9ybT0ibWF0cml4KDM0LjQyOTg2NywgMCwgMCwgMzQuNDI5ODY3LCAyMS44OTUzNTEsIDkwLjA5NzQwOSkiLz4KPHBhdGggZmlsbD0ibm9uZSIgc3Ryb2tlLXdpZHRoPSIwLjAzMzMzMzMiIHN0cm9rZS1saW5lY2FwPSJidXR0IiBzdHJva2UtbGluZWpvaW49Im1pdGVyIiBzdHJva2U9InJnYigwJSwgMCUsIDAlKSIgc3Ryb2tlLW9wYWNpdHk9IjEiIHN0cm9rZS1taXRlcmxpbWl0PSIxMCIgZD0iTSAyLjkxMzYyMSAxLjg5ODU2OCBMIDIuMDg2NTMyIDEuODk4NTY4ICIgdHJhbnNmb3JtPSJtYXRyaXgoMzQuNDI5ODY3LCAwLCAwLCAzNC40Mjk4NjcsIDIxLjg5NTM1MSwgOTAuMDk3NDA5KSIvPgo8cGF0aCBmaWxsPSJub25lIiBzdHJva2Utd2lkdGg9IjAuMDMzMzMzMyIgc3Ryb2tlLWxpbmVjYXA9ImJ1dHQiIHN0cm9rZS1saW5lam9pbj0ibWl0ZXIiIHN0cm9rZT0icmdiKDAlLCAwJSwgMCUpIiBzdHJva2Utb3BhY2l0eT0iMSIgc3Ryb2tlLW1pdGVybGltaXQ9IjEwIiBkPSJNIDIuMDA0ODQ0IDMuNDcyMTkyIEwgMS40OTk5NjggMi41OTc5MDYgIiB0cmFuc2Zvcm09Im1hdHJpeCgzNC40Mjk4NjcsIDAsIDAsIDM0LjQyOTg2NywgMjEuODk1MzUxLCA5MC4wOTc0MDkpIi8+CjxwYXRoIGZpbGw9Im5vbmUiIHN0cm9rZS13aWR0aD0iMC4wMzMzMzMzIiBzdHJva2UtbGluZWNhcD0iYnV0dCIgc3Ryb2tlLWxpbmVqb2luPSJtaXRlciIgc3Ryb2tlPSJyZ2IoMCUsIDAlLCAwJSkiIHN0cm9rZS1vcGFjaXR5PSIxIiBzdHJva2UtbWl0ZXJsaW1pdD0iMTAiIGQ9Ik0gMi4xMDEwNTQgMy4zMDU1MjcgTCAxLjY5MjUwMiAyLjU5NzkwNiAiIHRyYW5zZm9ybT0ibWF0cml4KDM0LjQyOTg2NywgMCwgMCwgMzQuNDI5ODY3LCAyMS44OTUzNTEsIDkwLjA5NzQwOSkiLz4KPHBhdGggZmlsbD0ibm9uZSIgc3Ryb2tlLXdpZHRoPSIwLjAzMzMzMzMiIHN0cm9rZS1saW5lY2FwPSJidXR0IiBzdHJva2UtbGluZWpvaW49Im1pdGVyIiBzdHJva2U9InJnYigwJSwgMCUsIDAlKSIgc3Ryb2tlLW9wYWNpdHk9IjEiIHN0cm9rZS1taXRlcmxpbWl0PSIxMCIgZD0iTSAyLjAwNDg0NCAxLjcyMzUwNiBMIDEuNDk1MjAzIDIuNjA2MTg4ICIgdHJhbnNmb3JtPSJtYXRyaXgoMzQuNDI5ODY3LCAwLCAwLCAzNC40Mjk4NjcsIDIxLjg5NTM1MSwgOTAuMDk3NDA5KSIvPgo8cGF0aCBmaWxsPSJub25lIiBzdHJva2Utd2lkdGg9IjAuMDMzMzMzMyIgc3Ryb2tlLWxpbmVjYXA9ImJ1dHQiIHN0cm9rZS1saW5lam9pbj0ibWl0ZXIiIHN0cm9rZT0icmdiKDAlLCAwJSwgMCUpIiBzdHJva2Utb3BhY2l0eT0iMSIgc3Ryb2tlLW1pdGVybGltaXQ9IjEwIiBkPSJNIDEuNTA5NjEyIDIuNTk3OTA2IEwgMC43MjAzMDQgMi41OTc5MDYgIiB0cmFuc2Zvcm09Im1hdHJpeCgzNC40Mjk4NjcsIDAsIDAsIDM0LjQyOTg2NywgMjEuODk1MzUxLCA5MC4wOTc0MDkpIi8+CjxwYXRoIGZpbGw9Im5vbmUiIHN0cm9rZS13aWR0aD0iMC4wMzMzMzMzIiBzdHJva2UtbGluZWNhcD0iYnV0dCIgc3Ryb2tlLWxpbmVqb2luPSJtaXRlciIgc3Ryb2tlPSJyZ2IoMCUsIDAlLCAwJSkiIHN0cm9rZS1vcGFjaXR5PSIxIiBzdHJva2UtbWl0ZXJsaW1pdD0iMTAiIGQ9Ik0gMC4zNDYzNTUgMi4zMzE3NCBMIDAuMTU3NzkyIDIuMDA1MjE2ICIgdHJhbnNmb3JtPSJtYXRyaXgoMzQuNDI5ODY3LCAwLCAwLCAzNC40Mjk4NjcsIDIxLjg5NTM1MSwgOTAuMDk3NDA5KSIvPgo8cGF0aCBmaWxsPSJub25lIiBzdHJva2Utd2lkdGg9IjAuMDMzMzMzMyIgc3Ryb2tlLWxpbmVjYXA9ImJ1dHQiIHN0cm9rZS1saW5lam9pbj0ibWl0ZXIiIHN0cm9rZT0icmdiKDAlLCAwJSwgMCUpIiBzdHJva2Utb3BhY2l0eT0iMSIgc3Ryb2tlLW1pdGVybGltaXQ9IjEwIiBkPSJNIDAuMzQ2MzU1IDIuODY0MDcyIEwgMC4xNTc3OTIgMy4xOTA0ODMgIiB0cmFuc2Zvcm09Im1hdHJpeCgzNC40Mjk4NjcsIDAsIDAsIDM0LjQyOTg2NywgMjEuODk1MzUxLCA5MC4wOTc0MDkpIi8+CjxnIGZpbGw9InJnYigxOSUsIDMxJSwgOTcuMDAwMDAzJSkiIGZpbGwtb3BhY2l0eT0iMSI+Cjx1c2UgeGxpbms6aHJlZj0iI2dseXBoLTAtMSIgeD0iMTg4Ljg5ODQzNyIgeT0iMTU0Ljc0NjA5NCIvPgo8L2c+CjxnIGZpbGw9InJnYigxMDAlLCA1LjElLCA1LjElKSIgZmlsbC1vcGFjaXR5PSIxIj4KPHVzZSB4bGluazpocmVmPSIjZ2x5cGgtMC0yIiB4PSIyMjMuMDU4NTk0IiB5PSI5NS4xMTMyODEiLz4KPC9nPgo8ZyBmaWxsPSJyZ2IoMTAwJSwgNS4xJSwgNS4xJSkiIGZpbGwtb3BhY2l0eT0iMSI+Cjx1c2UgeGxpbms6aHJlZj0iI2dseXBoLTAtMyIgeD0iMjEzLjY3OTY4OCIgeT0iOTQuOTI5Njg4Ii8+CjwvZz4KPGcgZmlsbD0icmdiKDE5JSwgMzElLCA5Ny4wMDAwMDMlKSIgZmlsbC1vcGFjaXR5PSIxIj4KPHVzZSB4bGluazpocmVmPSIjZ2x5cGgtMC0xIiB4PSIzMy45NTcwMzEiIHk9IjE4NC41NjI1Ii8+CjwvZz4KPGcgZmlsbD0icmdiKDAlLCAwJSwgMCUpIiBmaWxsLW9wYWNpdHk9IjEiPgo8dXNlIHhsaW5rOmhyZWY9IiNnbHlwaC0wLTQiIHg9IjE3LjA3NDIxOSIgeT0iMTU0Ljc0MjE4OCIvPgo8L2c+CjxnIGZpbGw9InJnYigwJSwgMCUsIDAlKSIgZmlsbC1vcGFjaXR5PSIxIj4KPHVzZSB4bGluazpocmVmPSIjZ2x5cGgtMC0zIiB4PSIyLjE0MDYyNSIgeT0iMTU0LjU1ODU5NCIvPgo8L2c+CjxnIGZpbGw9InJnYigwJSwgMCUsIDAlKSIgZmlsbC1vcGFjaXR5PSIxIj4KPHVzZSB4bGluazpocmVmPSIjZ2x5cGgtMS0xIiB4PSIxMS41OTM3NSIgeT0iMTU1LjM0NzY1NiIvPgo8L2c+CjxnIGZpbGw9InJnYigwJSwgMCUsIDAlKSIgZmlsbC1vcGFjaXR5PSIxIj4KPHVzZSB4bGluazpocmVmPSIjZ2x5cGgtMC00IiB4PSIxNy4wNzQyMTkiIHk9IjIxNC4zNzUiLz4KPC9nPgo8ZyBmaWxsPSJyZ2IoMCUsIDAlLCAwJSkiIGZpbGwtb3BhY2l0eT0iMSI+Cjx1c2UgeGxpbms6aHJlZj0iI2dseXBoLTAtMyIgeD0iMi4xNDA2MjUiIHk9IjIxNC4xOTE0MDYiLz4KPC9nPgo8ZyBmaWxsPSJyZ2IoMCUsIDAlLCAwJSkiIGZpbGwtb3BhY2l0eT0iMSI+Cjx1c2UgeGxpbms6aHJlZj0iI2dseXBoLTEtMSIgeD0iMTEuNTkzNzUiIHk9IjIxNC45ODA0NjkiLz4KPC9nPgo8L3N2Zz4K)
CAS
3230-43-1
化学式
C15H16N2O
mdl
——
分子量
240.305
InChiKey
GJZBNSJPZKCNPU-UHFFFAOYSA-N
BEILSTEIN
——
EINECS
——
-
物化性质
-
计算性质
-
ADMET
-
安全信息
-
SDS
-
制备方法与用途
-
上下游信息
-
文献信息
-
表征谱图
-
同类化合物
-
相关功能分类
-
相关结构分类
计算性质
-
辛醇/水分配系数(LogP):3
-
重原子数:18
-
可旋转键数:3
-
环数:2.0
-
sp3杂化的碳原子比例:0.13
-
拓扑面积:35.8
-
氢给体数:1
-
氢受体数:3
SDS
上下游信息
-
下游产品
中文名称 英文名称 CAS号 化学式 分子量 2-(苯基亚甲基氨基)苯酚 2-(benzylideneamino)phenol 3230-45-3 C13H11NO 197.236 —— 2-(4-cyanobenzylideneamino)phenol —— C14H10N2O 222.246 —— 2-(4-nitro-benzylidenamino)-phenol 841-14-5 C13H10N2O3 242.234 —— 2-(4-methoxybenzylideneamino)phenol 3117-67-7 C14H13NO2 227.263 2-羟基缩苯胺 2-[(2-hydroxyphenyl)iminomethyl]phenol 1761-56-4 C13H11NO2 213.236
反应信息
-
作为反应物:描述:参考文献:名称:Selective conversion of C=N bonds to their corresponding carbonyl compounds by the tribromoisocyanuric acid/wet SiO2 system as a novel reagent摘要:Tribromoisocyanuric acid/wet SiO2 was used for the conversion of C=N bonds to their corresponding carbonyl compounds in oximes, semicarbazones, azines, and Schiff bases. The interesting feature of this system is that in those oximes, semicarbazones, azines, and Schiff bases which have conjugated or unconjugated C=C bonds, the C=N bond will selectively change to the relevant C=O bond while the conjugated or unconjugated C=C bond will remain intact.DOI:10.1007/s00706-011-0649-5
-
作为产物:参考文献:名称:Co(II) 和 Cu(II) 混合配体配合物的合成、光谱表征、计算研究、NLO 特性和抗菌活性的理论研究摘要:摘要 席夫碱 2-(4-(二甲氨基)亚苄基氨基)苯酚 (DBAP) 和二亚乙基三胺/乙二胺与钴 (II) 和铜 (II) 金属离子以等摩尔比反应得到混合配体配合物 [M(DBAP)(L) Cl(H 2 O) n ] ( 1 – 4 ), 其中 M = Co(II) 1 , 2 ; Cu(II) 3 , 4 ; L = 二亚乙基三胺 ( 1 , 3 ); n = 0 和乙二胺 ( 2 , 4 ); n = 1。所有新合成的配合物均通过元素分析、光谱技术进行表征。、紫外-可见、FT-IR、ESR、PXRD和质谱。用DFT研究考察了钴(II)和铜(II)离子螯合对席夫碱(DBAP)非线性光学性质的影响。计算配体及其金属配合物的极化率(α 0)和一级超极化率(β),并与参考分子对硝基苯胺进行比较。席夫碱及其金属配合物的超极化表现出良好的非线性光学行为。配合物的理论研究通过 DFT 在基组 6-31DOI:10.1080/00958972.2021.2022128
文献信息
-
A DFT and experimental study of the spectroscopic and hydrolytic degradation behaviour of some benzylideneanilines作者:Tahjna I. Robertson、Peter N. NelsonDOI:10.1016/j.molstruc.2021.131625日期:2022.2The spectroscopic and hydrolytic degradation behaviour of some N-benzylideneanilines are investigated experimentally and theoretically via high quality density function theoretical (DFT) modelling techniques. Their absorption and vibrational spectra, accurately predicted by DFT calculations, are highly dependent on the nature of the substituents on the aromatic rings, hence, though some of their spectroscopic通过高质量密度函数理论 (DFT) 建模技术,通过实验和理论研究了一些N-亚苄基苯胺的光谱和水解降解行为。通过 DFT 计算准确预测它们的吸收和振动光谱高度依赖于芳环上取代基的性质,因此,尽管它们的一些光谱特征相似,但由于它们的电子结构不同,因此存在能量差异。而邻羟基苯胺衍生的加合物通过两种途径进行水解,其中最经济的途径是由快速焓驱动的水合引发,超过53 kJ mol -1的保守自由能 (ΔG ǂ ) 势垒,在通过约 132 kJ mol -1的保守屏障发生的限速熵控制裂解步骤之前,所有其他化合物通过较慢的两步途径水解,受水合步骤的限制。两种途径的势垒高度主要由结构控制,因此,过渡态的稳定性,所有这些对于两种途径都是循环的。
-
Preparation of β-Lactams by Mannich-Type Addition of Ethyl(trimethylsilyl)acetate (ETSA) to N-(2-Hydroxyphenyl)aldimine Sodium Salts作者:Sylvain Oudeyer、Vincent Levacher、Vincent Gembus、Thomas Poisson、Francis MarsaisDOI:10.1055/s-0029-1217729日期:2009.9The synthesis of highly substituted β-lactams was achieved by addition of air-stable ethyl(trimethylsilyl)acetate derivatives to N-(2-hydroxyphenyl)aldimine sodium salts in a THF-EtCN mixture. This reaction proceeds with moderate to good yields and diastereomeric ratio of up to 78:22. The reactivity of the N-(2-hydroxyphenyl)aldimine can be modified by simply changing the co-solvent from EtCN to MeCN to afford the cyanomethylated addition product.
-
Abnormal effect of hydroxyl on the longest wavelength maximum in ultraviolet absorption spectra for bis-aryl Schiff bases作者:Chao-Tun Cao、Wei Zhou、Chenzhong CaoDOI:10.1002/poc.3672日期:2017.104‐HOArCH=NArY and XArCH=NArOH‐4′, and the second set consists of 2‐HOArCH=NArY and XArCH=NArOH‐2′. Their ultraviolet absorption spectra were measured and investigated. A very interesting phenomenon was observed by analyzing their wave number νmax (cm−1) of longest wavelength maximum λmax (nm) of ultraviolet. Compared with the change regularity of the νmax of XArCH=NArY (where the X and Y excluded OH), the合成了两组包含4(或4')-OH和2(或2')-OH的双芳基席夫碱。第一组由4-HOArCH = NArY和XArCH = NArOH-4'组成,第二组由2-HOArCH = NArY和XArCH = NArOH-2'组成。测量并研究了它们的紫外线吸收光谱。通过分析它们的紫外线的最长波长最大值λmax(nm)的波数νmax(cm -1),观察到非常有趣的现象。与XArCH = NArY的νmax的变化规律相比(X和Y排除了OH),4'-位置羟基(4'-OH)和2'-位置羟基(2'-OH)具有异常的性能。详细信息如下:4'-OH对νmax产生额外的红移XArCH = NArOH-2'(λmax减小)的最大值,而2'-OH对XArCH = NArOH-2'的νmax贡献了额外的蓝移(λmax减小)。此外,对于2-HOArCH = NArY和XArCH = NArOH-2',所有2-OH和2'-
-
Heterochelates of ruthenium(II): electrochemistry, absorption spectra, and luminescence properties作者:Sujoy Baitalik、Bibhutosh AdhikaryDOI:10.1016/s0277-5387(97)00174-5日期:1997.9voltammograms of all the complexes, except6 (X = NMe2), exhibit a reversible RuII/RuIII and an irreversible RuIII/RuIV oxidation processes. In the case of6, both the oxidation processes take place reversibly. A linear correlation is obtained by plotting the E1/2 value of RuII/RuIII couple against the Hammett σ+ parameters. The absorption spectra of the complexes are similar showing absorption bands due to dπ-π*一系列heterochelates的的[Ru(联吡啶)2(LX)](CLO 4)(1-6)从的席夫碱衍生ö氨基苯酚和p-取代的(X)的苯甲醛,HL-X(X = NO 2,氯,F,Me,OMe和NMe 2)已经合成并表征。除6(X = NMe 2)外,所有配合物的循环伏安图均显示可逆的Ru II / Ru III和不可逆的Ru III / Ru IV氧化过程。在6的情况下,两个氧化过程均可逆地进行。通过绘制E来获得线性相关性Ru II / Ru III的1/2值与Hammettσ +参数耦合。复合物的吸收光谱相似表示吸收带由于dπ-π *(BPY),席夫碱Ñ -π *和π-π * intraligand过渡。在νmax(MLCT)和ΔE 1/2(第一氧化和第一还原对之间的间隔)之间也获得线性相关。在MLCT和溶剂的效果Ñ -π *已经研究了吸收带。荧光光谱研究表明,所有配合物在室温下发光,观
-
Oxidative NHC Catalysis for the Generation of Imidoyl Azoliums: Synthesis of Benzoxazoles作者:Atanu Patra、Anjima James、Tamal Kanti Das、Akkattu T. BijuDOI:10.1021/acs.joc.8b02598日期:2018.12.7N-Heterocyclic carbene (NHC)-catalyzed intramolecular cyclization of aldimines generated from 2-amino phenols and aromatic aldehydes leading to the synthesis of 2-arylbenzoxazoles under mild conditions is presented. The reaction proceeds via the generation of the aza-Breslow intermediates from imines and NHC, which under oxidative conditions form the key imidoyl azoliums and a subsequent intramolecular
表征谱图
-
氢谱1HNMR
-
质谱MS
-
碳谱13CNMR
-
红外IR
-
拉曼Raman
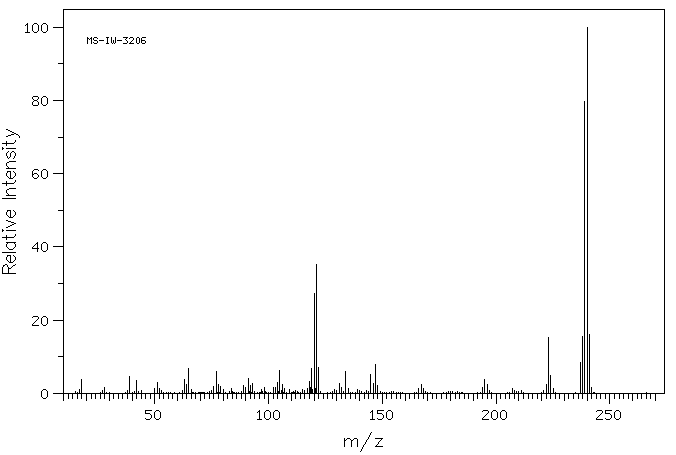
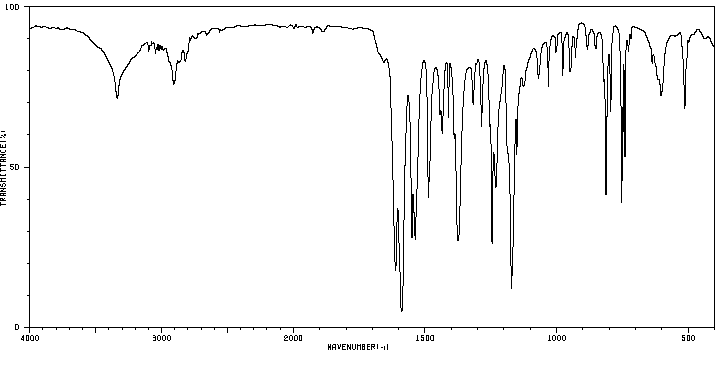
-
峰位数据
-
峰位匹配
-
表征信息
同类化合物
(乙腈)二氯镍(II)
(R)-(-)-α-甲基组胺二氢溴化物
(N-(2-甲基丙-2-烯-1-基)乙烷-1,2-二胺)
(4-(苄氧基)-2-(哌啶-1-基)吡啶咪丁-5-基)硼酸
(11-巯基十一烷基)-,,-三甲基溴化铵
鼠立死
鹿花菌素
鲸蜡醇硫酸酯DEA盐
鲸蜡硬脂基二甲基氯化铵
鲸蜡基胺氢氟酸盐
鲸蜡基二甲胺盐酸盐
高苯丙氨醇
高箱鲀毒素
高氯酸5-(二甲氨基)-1-({(E)-[4-(二甲氨基)苯基]甲亚基}氨基)-2-甲基吡啶正离子
高氯酸2-氯-1-({(E)-[4-(二甲氨基)苯基]甲亚基}氨基)-6-甲基吡啶正离子
高氯酸2-(丙烯酰基氧基)-N,N,N-三甲基乙铵
马诺地尔
马来酸氢十八烷酯
马来酸噻吗洛尔EP杂质C
马来酸噻吗洛尔
马来酸倍他司汀
顺式环己烷-1,3-二胺盐酸盐
顺式氯化锆二乙腈
顺式吡咯烷-3,4-二醇盐酸盐
顺式双(3-甲氧基丙腈)二氯铂(II)
顺式3,4-二氟吡咯烷盐酸盐
顺式1-甲基环丙烷1,2-二腈
顺式-二氯-反式-二乙酸-氨-环己胺合铂
顺式-二抗坏血酸(外消旋-1,2-二氨基环己烷)铂(II)水合物
顺式-N,2-二甲基环己胺
顺式-4-甲氧基-环己胺盐酸盐
顺式-4-环己烯-1.2-二胺
顺式-4-氨基-2,2,2-三氟乙酸环己酯
顺式-3-氨基环丁烷甲腈盐酸盐
顺式-2-羟基甲基-1-甲基-1-环己胺
顺式-2-甲基环己胺
顺式-2-(苯基氨基)环己醇
顺式-2-(苯基氨基)环己醇
顺式-2-(氨基甲基)-1-苯基环丙烷羧酸盐酸盐
顺式-1,3-二氨基环戊烷
顺式-1,2-环戊烷二胺二盐酸盐
顺式-1,2-环戊烷二胺
顺式-1,2-环丁腈
顺式-1,2-双氨甲基环己烷
顺式--N,N'-二甲基-1,2-环己二胺
顺式-(R,S)-1,2-二氨基环己烷铂硫酸盐
顺式-(2-氨基-环戊基)-甲醇
顺-2-戊烯腈
顺-1,3-环己烷二胺
顺-1,3-双(氨甲基)环己烷

联系我们



关注我们

公众号

