
3-苯基丙基硫代异氰酸酯 | 2627-27-2
中文名称
3-苯基丙基硫代异氰酸酯
中文别名
3-苯基异硫氰酸丙酯;3-苯基丙基异硫氰酸酯
英文名称
3-phenylpropyl isothiocyanate
英文别名
(3-isothiocyanatopropyl)benzene;3-Phenylpropylisothiocyanat;Phenylpropyl isothiocyanate;3-isothiocyanatopropylbenzene

CAS
2627-27-2
化学式
C10H11NS
mdl
——
分子量
177.27
InChiKey
GRUOGLPIAPZLHJ-UHFFFAOYSA-N
BEILSTEIN
——
EINECS
——
-
物化性质
-
计算性质
-
ADMET
-
安全信息
-
SDS
-
制备方法与用途
-
上下游信息
-
文献信息
-
表征谱图
-
同类化合物
-
相关功能分类
-
相关结构分类
物化性质
-
沸点:156 °C
-
密度:1.07
-
闪点:156-160°C/12mm
-
保留指数:1522
-
稳定性/保质期:
如果按照规格使用和储存,则不会分解。
计算性质
-
辛醇/水分配系数(LogP):3.9
-
重原子数:12
-
可旋转键数:4
-
环数:1.0
-
sp3杂化的碳原子比例:0.3
-
拓扑面积:44.4
-
氢给体数:0
-
氢受体数:2
安全信息
-
危险等级:8
-
危险类别码:R20/21/22,R34
-
危险品运输编号:2927
-
海关编码:2930909090
-
包装等级:III
-
危险类别:8
-
安全说明:S26,S36/37/39,S45
-
储存条件:密封于阴凉干燥处。
SDS
上下游信息
-
上游原料
中文名称 英文名称 CAS号 化学式 分子量 3-苯基-1-丙胺 3-Phenylpropan-1-amine 2038-57-5 C9H13N 135.209 -
下游产品
中文名称 英文名称 CAS号 化学式 分子量 1-(3-苯丙基)-2-硫脲 N-(3-Phenylpropyl)thiourea 93168-20-8 C10H14N2S 194.301
反应信息
-
作为反应物:描述:3-苯基丙基硫代异氰酸酯 在 三氯化铝 作用下, 以 various solvent(s) 为溶剂, 反应 1.0h, 以12%的产率得到1-Thiono-2,3,4,5-tetrahydro-1H-benz
azepin 参考文献:名称:脂氧合酶抑制剂,第 3 版:四氢苯并氮杂-苯腙的合成摘要:通过贝克曼重排或施密特反应获得了作为起始原料所需的四氢-2H-苯[b]氮杂-2-化合物。苯腙由硫酮和硫代内酰胺醚合成。测定了大豆脂氧化酶的抑制作用。DOI:10.1002/ardp.19943270208 -
作为产物:描述:Triethylammonium-3-phenylpropyldithiocarbamat 在 对甲苯磺酰氯 作用下, 以 四氢呋喃 为溶剂, 反应 1.0h, 以75%的产率得到3-苯基丙基硫代异氰酸酯参考文献:名称:甲苯磺酰氯的异硫氰酸盐介导原位生成的二硫代氨基甲酸盐的分解摘要:据报道,由烷基胺和芳基胺制备异硫氰酸酯的简便通用方法。该方法依赖于甲苯磺酰氯介导的二硫代氨基甲酸盐的分解,该二硫代氨基甲酸盐是通过用二硫化碳和三乙胺处理胺而原位产生的。利用该方案,我们以中等至极好的收率制备了19-烷基异硫氰酸酯和芳基异硫氰酸酯。DOI:10.1021/jo070246n
文献信息
-
Synthesis of Isothiocyanates and Unsymmetrical Thioureas with the Bench-Stable Solid Reagent (Me<sub>4</sub>N)SCF<sub>3</sub>作者:Thomas Scattolin、Alexander Klein、Franziska SchoenebeckDOI:10.1021/acs.orglett.7b00689日期:2017.4.7selective, and rapid transformation of primary amines and diamines to isothiocyanates and cyclic thioureas is disclosed. As opposed to established approaches that employ toxic or volatile electrophilic liquids and require reaction control (i.e., slow addition, cooling), this protocol utilizes the bench-stable, solid reagent (Me4N)SCF3 at room temperature. The method is characterized by operational simplicity
-
Reaction of Thiocarbonyl Fluoride Generated from Difluorocarbene with Amines作者:Jiao Yu、Jin-Hong Lin、Ji-Chang XiaoDOI:10.1002/anie.201710186日期:2017.12.22The reaction of thiocarbonyl fluoride, generated from difluorocarbene, with various amines under mild conditions is described. Secondary amines, primary amines, and o‐phenylenediamines are converted to thiocarbamoyl fluorides, isothiocyanates, and difluoromethylthiolated heterocycles, respectively. Thiocarbamoyl fluorides were further transformed into trifluoromethylated amines by using a one‐pot process
-
T3P® – A Benign Desulfurating Reagent in the Synthesis of Isothiocyanates作者:Tadeusz Gajda、Łukasz Janczewski、Anna Gajda、Sebastian Frankowski、Tomasz GoszczyńskiDOI:10.1055/s-0036-1591842日期:2018.3Abstract A number of alkyl, aryl and bifunctional isothiocyanates are obtained in moderate to high yields (41–94%) in a two-step, one-pot reaction of the parent primary amines or their salts with carbon disulfide, followed by reaction of the thus formed dithiocarbamates with T3P® (propane phosphonic acid anhydride) as a new and efficient desulfurating agent. A number of alkyl, aryl and bifunctional isothiocyanates
-
Discovery of novel Methylsulfonyl phenyl derivatives as potent human Cyclooxygenase-2 inhibitors with effective anticonvulsant action: Design, synthesis, in-silico, in-vitro and in-vivo evaluation作者:Chandra Bhushan Mishra、Shikha Kumari、Amresh Prakash、Rajesh Yadav、Ankit Kumar Tiwari、Preeti Pandey、Manisha TiwariDOI:10.1016/j.ejmech.2018.04.007日期:2018.5seizure test and MTL-1 excellently protected animals against PTZ induced seizure at the dose of 30 mg/kg. MTL-1 also indicates long duration of action in time course study and displayed significant seizure protection up to 6 h of drug administration. Further, the anti-epileptogenic effect of MTL-1 has been examined in PTZ induced chronic model of epilepsy. The results indicated that MTL-1 had a significant设计并合成了一系列新的甲基磺酰基苯基衍生物,以评估其对COX-2的抑制活性以及抗惊厥潜能。体外评估表明,两种化合物MTL-1和MTL-2似乎是整个系列中最有效和选择性的COX-2抑制剂。在sc-PTZ诱发的癫痫发作试验中评估了两种有效的COX-2抑制剂的抗惊厥活性,MTL-1以30 mg / kg的剂量出色地保护了动物免受PTZ诱发的癫痫发作。MTL-1在时程研究中还表明作用时间长,在给药6小时后显示出明显的癫痫发作保护作用。此外,已经在PTZ诱导的癫痫慢性模型中检查了MTL-1的抗癫痫作用。结果表明,与Etoricoxib(ETX)和PTZ单独治疗组相比,MTL-1在PTZ点燃的大鼠中具有显着的抗癫痫发生作用。此外,MTL-1成功改善了PTZ点燃的大鼠的认知功能障碍,这已得到社会认可,新颖的物体识别和明暗室测试的证实。此外,还进行了分子对接和分子模拟(MD模拟)研究,以阐明MTL-1与C
-
Identification of a New Heterocyclic Scaffold for Inhibitors of the Polo-Box Domain of Polo-like Kinase 1作者:Celeste N. Alverez、Jung-Eun Park、Kiran S. Toti、Yangliu Xia、Kristopher W. Krausz、Ganesha Rai、Jeong K. Bang、Frank J. Gonzalez、Kenneth A. Jacobson、Kyung S. LeeDOI:10.1021/acs.jmedchem.0c01669日期:2020.11.25As a mitotic-specific target widely deregulated in various human cancers, polo-like kinase 1 (Plk1) has been extensively explored for anticancer activity and drug discovery. Although multiple catalytic domain inhibitors were tested in preclinical and clinical studies, their efficacies are limited by dose-limiting cytotoxicity, mainly from off-target cross reactivity. The C-terminal noncatalytic polo-box作为在各种人类癌症中广泛失调的有丝分裂特异性靶标,polo 样激酶 1 (Plk1) 已被广泛探索用于抗癌活性和药物发现。尽管在临床前和临床研究中测试了多种催化结构域抑制剂,但它们的功效受到剂量限制性细胞毒性的限制,主要来自脱靶交叉反应。 Plk1 的 C 端非催化 polo-box 结构域 (PBD) 已成为产生新的蛋白质-蛋白质相互作用抑制剂的有吸引力的靶标。在这里,我们鉴定了一种 1-thioxo-2,4-dihydro-[1,2,4]triazolo[4,3 -a ]quinazolin-5(1 H )-one 支架,可有效抑制 Plk1 PBD,但不能抑制其相关的 Plk2和 Plk3 PBD。结构-活性关系研究发现多种抑制剂的抑制活性比之前表征的 Plk1 PBD 特异性磷酸肽 PLHSpT ( K d ∼ 450 nM) 高 ≥10 倍。此外, S-甲基前药有效抑制有丝分裂进
表征谱图
-
氢谱1HNMR
-
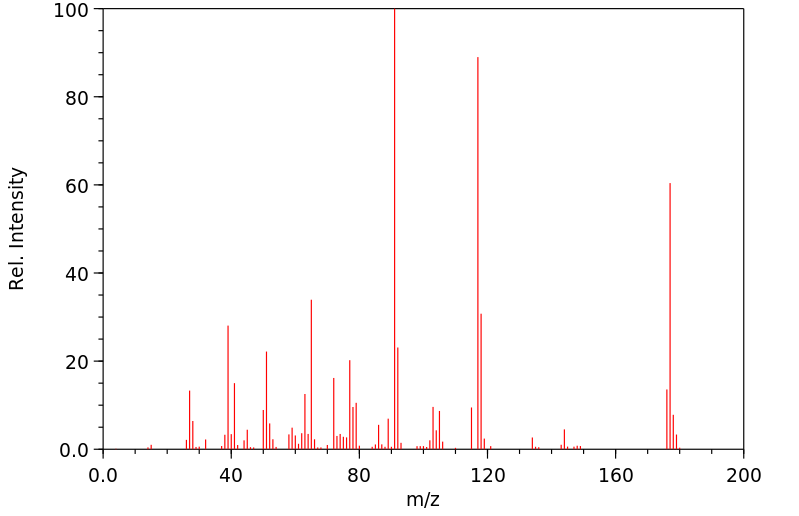
质谱MS
-
碳谱13CNMR
-
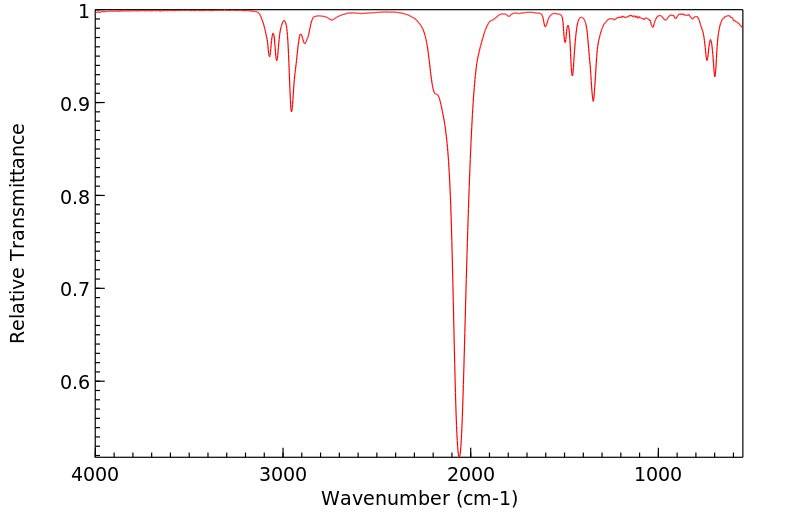
红外IR
-
拉曼Raman
-
峰位数据
-
峰位匹配
-
表征信息
同类化合物
(βS)-β-氨基-4-(4-羟基苯氧基)-3,5-二碘苯甲丙醇
(S,S)-邻甲苯基-DIPAMP
(S)-(-)-7'-〔4(S)-(苄基)恶唑-2-基]-7-二(3,5-二-叔丁基苯基)膦基-2,2',3,3'-四氢-1,1-螺二氢茚
(S)-盐酸沙丁胺醇
(S)-3-(叔丁基)-4-(2,6-二甲氧基苯基)-2,3-二氢苯并[d][1,3]氧磷杂环戊二烯
(S)-2,2'-双[双(3,5-三氟甲基苯基)膦基]-4,4',6,6'-四甲氧基联苯
(S)-1-[3,5-双(三氟甲基)苯基]-3-[1-(二甲基氨基)-3-甲基丁烷-2-基]硫脲
(R)富马酸托特罗定
(R)-(-)-盐酸尼古地平
(R)-(-)-4,12-双(二苯基膦基)[2.2]对环芳烷(1,5环辛二烯)铑(I)四氟硼酸盐
(R)-(+)-7-双(3,5-二叔丁基苯基)膦基7''-[((6-甲基吡啶-2-基甲基)氨基]-2,2'',3,3''-四氢-1,1''-螺双茚满
(R)-(+)-7-双(3,5-二叔丁基苯基)膦基7''-[(4-叔丁基吡啶-2-基甲基)氨基]-2,2'',3,3''-四氢-1,1''-螺双茚满
(R)-(+)-7-双(3,5-二叔丁基苯基)膦基7''-[(3-甲基吡啶-2-基甲基)氨基]-2,2'',3,3''-四氢-1,1''-螺双茚满
(R)-(+)-4,7-双(3,5-二-叔丁基苯基)膦基-7“-[(吡啶-2-基甲基)氨基]-2,2”,3,3'-四氢1,1'-螺二茚满
(R)-3-(叔丁基)-4-(2,6-二苯氧基苯基)-2,3-二氢苯并[d][1,3]氧杂磷杂环戊烯
(R)-2-[((二苯基膦基)甲基]吡咯烷
(R)-1-[3,5-双(三氟甲基)苯基]-3-[1-(二甲基氨基)-3-甲基丁烷-2-基]硫脲
(N-(4-甲氧基苯基)-N-甲基-3-(1-哌啶基)丙-2-烯酰胺)
(5-溴-2-羟基苯基)-4-氯苯甲酮
(5-溴-2-氯苯基)(4-羟基苯基)甲酮
(5-氧代-3-苯基-2,5-二氢-1,2,3,4-oxatriazol-3-鎓)
(4S,5R)-4-甲基-5-苯基-1,2,3-氧代噻唑烷-2,2-二氧化物-3-羧酸叔丁酯
(4S,4''S)-2,2''-亚环戊基双[4,5-二氢-4-(苯甲基)恶唑]
(4-溴苯基)-[2-氟-4-[6-[甲基(丙-2-烯基)氨基]己氧基]苯基]甲酮
(4-丁氧基苯甲基)三苯基溴化磷
(3aR,8aR)-(-)-4,4,8,8-四(3,5-二甲基苯基)四氢-2,2-二甲基-6-苯基-1,3-二氧戊环[4,5-e]二恶唑磷
(3aR,6aS)-5-氧代六氢环戊基[c]吡咯-2(1H)-羧酸酯
(2Z)-3-[[(4-氯苯基)氨基]-2-氰基丙烯酸乙酯
(2S,3S,5S)-5-(叔丁氧基甲酰氨基)-2-(N-5-噻唑基-甲氧羰基)氨基-1,6-二苯基-3-羟基己烷
(2S,2''S,3S,3''S)-3,3''-二叔丁基-4,4''-双(2,6-二甲氧基苯基)-2,2'',3,3''-四氢-2,2''-联苯并[d][1,3]氧杂磷杂戊环
(2S)-(-)-2-{[[[[3,5-双(氟代甲基)苯基]氨基]硫代甲基]氨基}-N-(二苯基甲基)-N,3,3-三甲基丁酰胺
(2S)-2-[[[[[((1S,2S)-2-氨基环己基]氨基]硫代甲基]氨基]-N-(二苯甲基)-N,3,3-三甲基丁酰胺
(2S)-2-[[[[[[((1R,2R)-2-氨基环己基]氨基]硫代甲基]氨基]-N-(二苯甲基)-N,3,3-三甲基丁酰胺
(2-硝基苯基)磷酸三酰胺
(2,6-二氯苯基)乙酰氯
(2,3-二甲氧基-5-甲基苯基)硼酸
(1S,2S,3S,5S)-5-叠氮基-3-(苯基甲氧基)-2-[(苯基甲氧基)甲基]环戊醇
(1S,2S,3R,5R)-2-(苄氧基)甲基-6-氧杂双环[3.1.0]己-3-醇
(1-(4-氟苯基)环丙基)甲胺盐酸盐
(1-(3-溴苯基)环丁基)甲胺盐酸盐
(1-(2-氯苯基)环丁基)甲胺盐酸盐
(1-(2-氟苯基)环丙基)甲胺盐酸盐
(1-(2,6-二氟苯基)环丙基)甲胺盐酸盐
(-)-去甲基西布曲明
龙蒿油
龙胆酸钠
龙胆酸叔丁酯
龙胆酸
龙胆紫-d6
龙胆紫

联系我们



关注我们

公众号

