
3-(4-hydroxy-4-phenylpiperidin-1-yl)propionitrile | 5193-57-7
中文名称
——
中文别名
——
英文名称
3-(4-hydroxy-4-phenylpiperidin-1-yl)propionitrile
英文别名
3-(4-hydroxy-4-phenylpiperidin-1-yl)propanenitrile;1-(Beta-cyanoethyl)-4-phenyl-4-piperidinol

CAS
5193-57-7
化学式
C14H18N2O
mdl
——
分子量
230.31
InChiKey
YFHOEXZVEVBIHH-UHFFFAOYSA-N
BEILSTEIN
——
EINECS
——
-
物化性质
-
计算性质
-
ADMET
-
安全信息
-
SDS
-
制备方法与用途
-
上下游信息
-
文献信息
-
表征谱图
-
同类化合物
-
相关功能分类
-
相关结构分类
物化性质
-
熔点:87-89 °C
-
沸点:432.1±45.0 °C(Predicted)
-
密度:1.125±0.06 g/cm3(Predicted)
计算性质
-
辛醇/水分配系数(LogP):1.1
-
重原子数:17
-
可旋转键数:3
-
环数:2.0
-
sp3杂化的碳原子比例:0.5
-
拓扑面积:47.3
-
氢给体数:1
-
氢受体数:3
上下游信息
-
上游原料
中文名称 英文名称 CAS号 化学式 分子量 4-苯基-4-羟基哌啶 4-hydroxy-4-phenylpiperidin 40807-61-2 C11H15NO 177.246 -
下游产品
中文名称 英文名称 CAS号 化学式 分子量 1-(3-氨基丙基)-4-羟基-4-苯基哌啶 1-(3-aminopropyl)-4-hydroxy-4-phenylpiperidine 5193-58-8 C14H22N2O 234.341
反应信息
-
作为反应物:描述:3-(4-hydroxy-4-phenylpiperidin-1-yl)propionitrile 在 盐酸 、 sodium hydroxide 作用下, 以 四氢呋喃 、 乙醚 、 二氯甲烷 、 水 为溶剂, 以3.12 g (83%)的产率得到1-(3-氨基丙基)-4-羟基-4-苯基哌啶参考文献:名称:Dihydropyridines and new uses thereof摘要:该发明提供了一种治疗良性前列腺增生的方法,包括向受试者施用具有以下结构的化合物的治疗有效量: 其中Y为--(CH.sub.2).sub.n --,其中n为1、2、3、4或5;--(CH.sub.2).sub.h --O--(CH.sub.2).sub.k --,其中h和k独立且相同或不同,为2、3或4;--(CH.sub.2).sub.h --CH.dbd.CH--(CH.sub.2).sub.k --;或--(CH.sub.2).sub.h --C.tbd.C--(CH.sub.2).sub.k --,其中h和k独立且相同或不同,为1、2、3或4;其中Z为O、NH或CH.sub.2;其中R.sup.1为线性或支链烷基、烷氧基烷基或芳基烷基;其中R.sup.2和R.sup.4独立且相同或不同,为H,或线性或支链烷基;其中R.sup.3为H、线性或支链烷基、烷氧基、烷氧基烷基或酰基;其中R.sup.5和R.sup.6独立且相同或不同,为H、OH、Cl、Br、F、NO.sub.2、CN、CF.sub.3或NH.sub.2,或线性或支链烷基、烷氧基、烷氧羰基、酰基、烷基亚砜、烷基砜、或单烷基或二烷基氨基基团。还公开了含有一、两个或三个环的其他活性化合物,以及由此制备的药物组合物和在治疗BPH、抑制胆固醇合成和降低眼压中的使用方法。公开号:US05767131A1
-
作为产物:描述:4-苯基-4-羟基哌啶 、 丙烯腈 以 乙醇 为溶剂, 反应 1.5h, 以to afford 3.71 g (92%) of white solid, which的产率得到3-(4-hydroxy-4-phenylpiperidin-1-yl)propionitrile参考文献:名称:Dihydropyridines and new uses thereof摘要:本发明提供了一种治疗良性前列腺增生的方法,该方法包括向受试者施用具有以下结构的化合物的治疗有效量:##STR1## 其中,Y为--(CH.sub.2).sub.n --,其中n为1、2、3、4或5;--(CH.sub.2).sub.h --O--(CH.sub.2).sub.k --,其中h和k独立且相同或不同,为2、3或4;--(CH.sub.2).sub.h --CH.dbd.CH--(CH.sub.2).sub.k --;或--(CH.sub.2).sub.h --C.tbd.C--(CH.sub.2).sub.k --,其中h和k独立且相同或不同,为1、2、3或4;其中Z为O、NH或CH.sub.2;其中R.sup.1为线性或支链烷基、烷氧基烷基或芳基烷基;其中R.sup.2和R.sup.4独立且相同或不同,为H或线性或支链烷基;其中R.sup.3为H、线性或支链烷基、烷氧基、烷氧基烷基或酰基;其中R.sup.5和R.sup.6独立且相同或不同,为H、OH、Cl、Br、F、NO.sub.2、CN、CF.sub.3或NH.sub.2,或线性或支链烷基、烷氧基、烷氧羰基、酰基、烷基亚砜、烷基磺酸、或单烷基或二烷基氨基基团。还公开了其他含有一、两个或三个环的活性化合物,以及制备自中的制药组合物和在治疗BPH、抑制胆固醇合成和降低眼内压力方面的使用方法。公开号:US05767131A1
文献信息
-
Bio-heterogeneous Cu(0)NC@PHA for n-aryl/alkylation at room temperature作者:Choong Jian Fui、Tang Xin Ting、Mohd Sani Sarjadi、Shaheen M. Sarkar、Baba Musta、Md Lutfor RahmanDOI:10.1016/j.poly.2021.115310日期:2021.9
-
A Structure−Activity Relationship Study of Novel Phenylacetamides Which Are Sodium Channel Blockers作者:Ioannis Roufos、Sheryl Hays、Roy D. SchwarzDOI:10.1021/jm950467y日期:1996.1.1A structure-activity relationship study of a series of novel Na+ channel blockers, structurally related to N-[3-(2,6-dimethyl-1-piperidinyl)propyl]-alpha-phenylbenzeneacetamide (1, PD85639) is described. The diphenylacetic acid portion of the molecule was left unchanged throughout the study, while structural features in the amine portion and the amide alkyl linkage of the molecule were modified. The compounds were tested for inhibition of veratridine-stimulated Na+ influx in CHO cells expressing type IIA Na+ channels. Several derivatives show a trend toward more potent Na+ channel blockade activity with increasing lipophilicity of the amine portion of the molecule. The presence of a phenyl ring near the amine increases inhibitory potency. A three-carbon spacer between the amide and amine is optimal, and a secondary amide linkage is preferred.
-
Design and Synthesis of Novel α<sub>1a</sub> Adrenoceptor-Selective Dihydropyridine Antagonists for the Treatment of Benign Prostatic Hyperplasia作者:Dhanapalan Nagarathnam、John M. Wetzel、Shou Wu Miao、Mohammad R. Marzabadi、George Chiu、Wai C. Wong、Xingfang Hong、James Fang、Carlos Forray、Theresa A. Branchek、William E. Heydorn、Raymond S. L. Chang、Theodore Broten、Terry W. Schorn、Charles GluchowskiDOI:10.1021/jm980506g日期:1998.12.1We report the synthesis and evaluation of novel alpha(1a) adrenoceptor subtype-selective antagonists. Systematic modification of the lipophilic 4,4-diphenylpiperidinyl moiety of the dihydropyridine derivatives 1 and 2 provided several highly selective and potent alpha(1a), antagonists. From this series, we identified the 4-(methoxycarbonyl)-4-phenylpiperidine analogue SNAP 5540 (-) [(-)-63] for further characterization. When examined in an isolated human prostate tissue assay, this compound was found to have a K-i of 2.8 nM, in agreement with the cloned human receptor binding data (K-i = 2.42 nM). Further evaluation of the compound in isolated dog prostate tissue showed a K-i of 3.6 nM and confirmed it to be a potent antagonist (K-b = 1.6 nM). In vivo, this compound effectively blocked the phenylephrine-stimulated increase in intraurethral pressure (IUP) in mongrel dogs, at doses which did not significantly affect the arterial pressure (diastolic blood pressure, DBP), with a DBP K-b/IUP K-b ratio of 16. In addition, (-)-63 also showed greater than 40 000-fold selectivity over the rat L-type calcium channel and 200-fold selectivity over several G protein-coupled receptors, including histamine and serotonin subtypes. These findings prove that alpha(1a) adrenoceptor-subtype selective antagonists such as (-)-63 may be developed as uroselective agents for an improved treatment of BPH over nonselective alpha(1) antagonists such as prazosin and terazosin, with fewer side effects.
-
US5767131A申请人:——公开号:US5767131A公开(公告)日:1998-06-16
-
US6211198B1申请人:——公开号:US6211198B1公开(公告)日:2001-04-03
表征谱图
-
氢谱1HNMR
-
质谱MS
-
碳谱13CNMR
-
红外IR
-
拉曼Raman
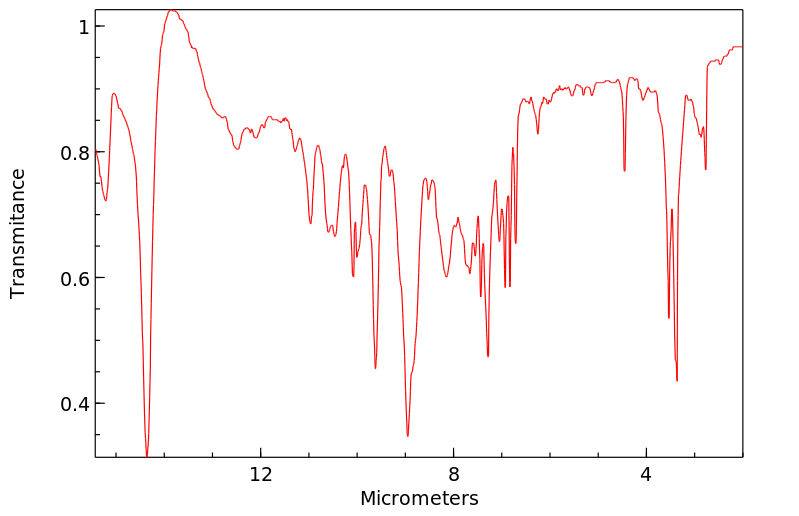
-
峰位数据
-
峰位匹配
-
表征信息
同类化合物
(R)-3-甲基哌啶盐酸盐;
(R)-2-苄基哌啶-1-羧酸叔丁酯
((3S,4R)-3-氨基-4-羟基哌啶-1-基)(2-(1-(环丙基甲基)-1H-吲哚-2-基)-7-甲氧基-1-甲基-1H-苯并[d]咪唑-5-基)甲酮盐酸盐
高氯酸哌啶
高托品酮肟
马来酸帕罗西汀
颜料红48:4
顺式3-氟哌啶-4-醇盐酸盐
顺式2,6-二甲基哌啶-4-酮
顺式1-苄基-4-甲基-3-甲氨基-哌啶
顺式-叔丁基4-羟基-3-甲基哌啶-1-羧酸酯
顺式-6-甲基-哌啶-1,3-二甲酸1-叔丁酯
顺式-5-(三氟甲基)哌啶-3-羧酸甲酯盐酸盐
顺式-4-叔丁基-2-甲基哌啶
顺式-4-Boc-氨基哌啶-3-甲酸甲酯
顺式-4-(氮杂环丁烷-1-基)-3-氟哌
顺式-3-顺式-4-氨基哌啶
顺式-3-甲氧基-4-氨基哌啶
顺式-3-BOC-3,7-二氮杂双环[4.2.0]辛烷
顺式-3-(1-吡咯烷基)环丁腈
顺式-3,5-哌啶二羧酸
顺式-3,4-二溴-3-甲基吡咯烷盐酸盐
顺式-2,6-二甲基-4-氧代哌啶-1-羧酸叔丁基酯
顺式-1-叔丁氧羰基-4-甲基氨基-3-羟基哌啶
顺式-1-boc-3,4-二氨基哌啶
顺式-1-(4-叔丁基环己基)-4-苯基-4-哌啶腈
顺式-1,3-二甲基-4-乙炔基-6-苯基-3,4-哌啶二醇
顺-4-(4-氟苯基)-1-(4-异丙基环己基)-4-哌啶羧酸
顺-4-(2-氟苯基)-1-(4-异丙基环己基)-4-哌啶羧酸
顺-3-氨基-4-氟哌啶-1-羧酸叔丁酯
顺-1-苄基-4-甲基哌啶-3-氨基酸甲酯盐酸盐
非莫西汀
雷芬那辛
雷拉地尔
阿维巴坦中间体4
阿格列汀杂质
阿尼利定盐酸盐 CII
阿尼利定
阿塔匹酮
阿哌沙班杂质BMS-591455
阿哌沙班杂质87
阿哌沙班杂质52
阿哌沙班杂质51
阿哌沙班杂质5
阿哌沙班杂质
阿哌沙班杂质
阿哌沙班-d3
阿哌沙班
阻聚剂701
间氨基谷氨酰胺

联系我们



关注我们

公众号

