
邻氨基苯甲酸戊酯 | 30100-15-3
中文名称
邻氨基苯甲酸戊酯
中文别名
——
英文名称
anthranilic acid n-pentyl ester
英文别名
pentyl 2-aminobenzoate;amyl anthranilate;n-pentyl anthranilate;Pentyl-anthranilat;Pentyl anthranilate

CAS
30100-15-3
化学式
C12H17NO2
mdl
——
分子量
207.272
InChiKey
JCKCYPSMCQDSHT-UHFFFAOYSA-N
BEILSTEIN
——
EINECS
——
-
物化性质
-
计算性质
-
ADMET
-
安全信息
-
SDS
-
制备方法与用途
-
上下游信息
-
文献信息
-
表征谱图
-
同类化合物
-
相关功能分类
-
相关结构分类
物化性质
-
沸点:312.9±15.0 °C(Predicted)
-
密度:1.059±0.06 g/cm3(Predicted)
-
LogP:4.217 (est)
-
保留指数:1700
计算性质
-
辛醇/水分配系数(LogP):4
-
重原子数:15
-
可旋转键数:6
-
环数:1.0
-
sp3杂化的碳原子比例:0.42
-
拓扑面积:52.3
-
氢给体数:1
-
氢受体数:3
安全信息
-
海关编码:2922439000
SDS
上下游信息
反应信息
-
作为反应物:描述:邻氨基苯甲酸戊酯 、 3,4-二甲氧基苯甲酰氯 在 三乙胺 作用下, 以 苯 为溶剂, 反应 3.0h, 生成 pentyl 2-[(3,4-dimethoxybenzoyl)amino]benzoate参考文献:名称:Structure-activity relationships of fungicidal N-benzoylanthranilic esters.摘要:37种N-(甲氧基取代的苯甲酰)蒽醌酯的抗真菌活性通过盆栽试验在由小麦白粉病(Erysiphe graminis)引起的大麦粉状霉菌上进行了测试。在测试的甲基N-(甲氧基取代的苯甲酰)蒽醌酯中,3,4-二甲氧基苯甲酰衍生物表现出最高的活性。N-(3,4-二甲氧基苯甲酰)蒽醌酯的杀真菌活性的变化被证明与酯的醇基的疏水性和电子性质的变化相关。醇基在α位的支链对活性是有害的。DOI:10.1271/bbb1961.44.2149
-
作为产物:描述:3-羟基-1,2,3-苯并三嗪-4(3H)-酮 以94%的产率得到参考文献:名称:AHERN T. P.; NAVRATIL T.; VAUGHAN K., CAN. J. CHEM.
, 1977, 55, NO 4, 630-639 摘要:DOI:
文献信息
-
Halogen-substituted anthranilic acid derivatives provide a novel chemical platform for androgen receptor antagonists作者:Daniela Roell、Thomas W. Rösler、Wiebke Hessenkemper、Florian Kraft、Monique Hauschild、Sophie Bartsch、Tsion E. Abraham、Adriaan B. Houtsmuller、Rudolf Matusch、Martin E. van Royen、Aria BaniahmadDOI:10.1016/j.jsbmb.2018.12.005日期:2019.4Androgen receptor (AR) antagonists are used for hormone therapy of prostate cancer (PCa). However resistance to the treatment occurs eventually. One possible reason is the occurrence of AR mutations that prevent inhibition of AR-mediated transactivation by antagonists. To offer in future more options to inhibit AR signaling, novel chemical lead structures for new AR antagonists would be beneficial. Here雄激素受体(AR)拮抗剂用于前列腺癌(PCa)的激素治疗。然而,最终对治疗产生抵抗力。一个可能的原因是,AR突变的发生阻止了拮抗剂对AR介导的反式激活的抑制。为了将来提供更多抑制AR信号传导的选择,新型AR拮抗剂的新型化学前导结构将是有益的。在这里,我们分析了36种邻氨基苯甲酸甲酯的非甾体结构变体,包括23种合成化合物的电池的结构活性关系。我们确定了导致更有效的AR拮抗剂的结构要求。特定的化合物抑制野生型AR的反式激活以及使突变体对羟基氟他胺,比卡鲁胺和第二代AR拮抗剂enzalutamide产生抗药性的AR突变体。这表明与临床使用的化合物相比,抑制AR的独特模式。竞争测定表明这些化合物与AR配体结合域结合并抑制PCa细胞增殖。而且,尽管抑制了AR介导的反式激活,这表明活性化合物不依赖于反式激活的AR途径,但是活性化合物诱导细胞衰老。与此相符,光漂白后的荧光共振(FRAP)-分析显示AR在
-
Process for preparing anthranilic acid esters
-
Compositions for use in golf balls申请人:Wu Shenshen公开号:US20050119451A1公开(公告)日:2005-06-02Golf balls comprising elastomer compositions are presently disclosed. These elastomer compositions comprise reaction products of polyisocyanates and telechelic polymers having isocyanate-reactive end-groups such as hydroxyl groups and/or amine groups. These elastomer compositions can be used in any one or more portions of the golf balls, such as inner core layer, intermediate core layer, outer core layer, intermediate layer, cover, inner cover layer, intermediate cover layer, and/or outer cover layer.
-
COMPOSITIONS FOR USE IN GOLF BALLS申请人:Rajagopalan Murali公开号:US20060014923A1公开(公告)日:2006-01-19A golf ball having a core and at least one layer disposed about the core is disclosed. The at least one layer is formed from a composition having multiple reactive and/or non-reactive ingredients. At least one of these ingredients is a sterically hindered polyamine, preferably formed from an active hydrogen functional compound and an amine/acid ortho- or meta-substituted cyclic compound.本发明公开了一种高尔夫球,它具有一个球芯和围绕球芯设置的至少一层。至少一层由具有多种活性和/或非活性成分的组合物形成。这些成分中至少有一种是立体受阻多胺,最好由活性氢官能团化合物和胺/酸正代或偏代环状化合物形成。
-
Photobleaching Activity of 2-(Phenylamino)methylidenecyclohexane-1,3-diones in Tobacco (<i>Nicotiana</i> <i>tabacum</i>) Cultured Cells作者:Jing-Ming Wang、Tadao Asami、Fan-Sik Che、Noboru Murofushi、Shigeo YoshidaDOI:10.1021/jf960962i日期:1997.7.1A series of phenylalkylidenecyclohexane-1,3-dione derivatives were designed and synthesized as vinylogous analogs of phthalimides, which are known photobleaching herbicides. The bleaching activity of synthesized compounds was assayed with photomixotrophic tobacco cultured cells under either light or dark conditions. Both the chlorophyll and the carotenoid contents of the cells treated with the cyclic diones decreased to almost zero within 24 h under the light condition but not under the dark condition. This rapid emergence of bleaching activity with light is one of the most distinctive actions of Protox-inhibiting herbicides; however, the cyclic dione did not inhibit protoporphyrinogen oxidase in vitro. Thus, we concluded that the cyclic diones possessed a different herbicidal mode of action from Protox-inhibiting herbicide.
表征谱图
-
氢谱1HNMR
-
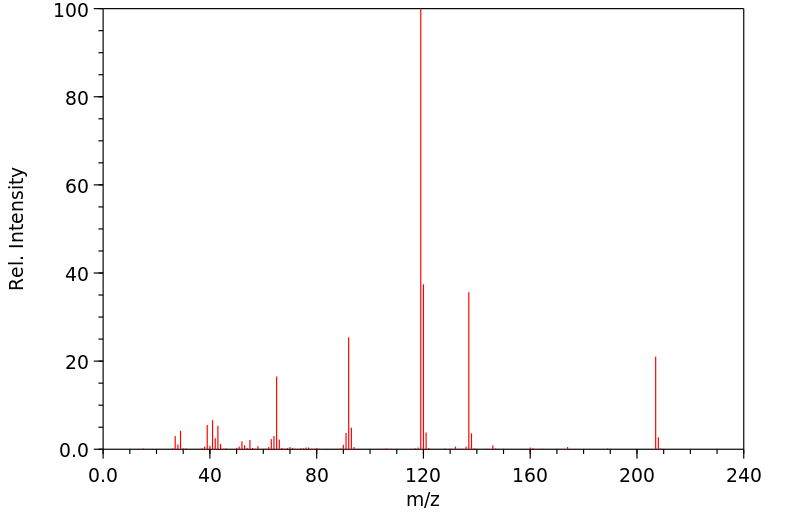
质谱MS
-
碳谱13CNMR
-
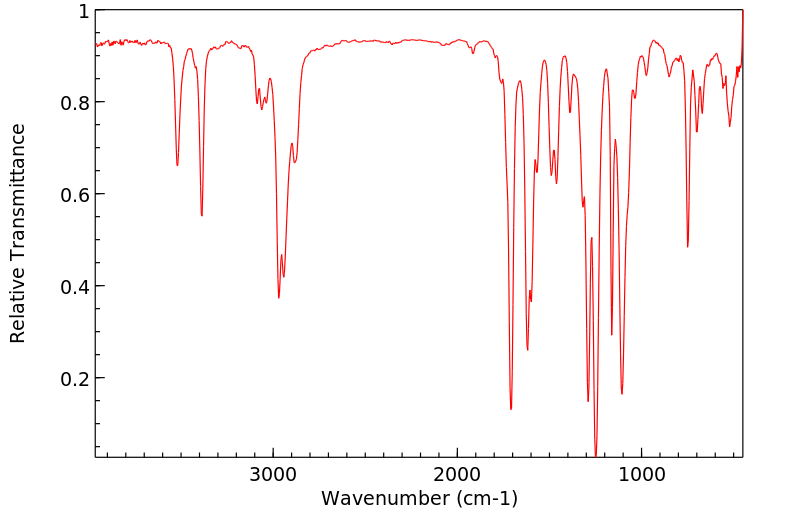
红外IR
-
拉曼Raman
-
峰位数据
-
峰位匹配
-
表征信息
同类化合物
(βS)-β-氨基-4-(4-羟基苯氧基)-3,5-二碘苯甲丙醇
(S,S)-邻甲苯基-DIPAMP
(S)-(-)-7'-〔4(S)-(苄基)恶唑-2-基]-7-二(3,5-二-叔丁基苯基)膦基-2,2',3,3'-四氢-1,1-螺二氢茚
(S)-盐酸沙丁胺醇
(S)-3-(叔丁基)-4-(2,6-二甲氧基苯基)-2,3-二氢苯并[d][1,3]氧磷杂环戊二烯
(S)-2,2'-双[双(3,5-三氟甲基苯基)膦基]-4,4',6,6'-四甲氧基联苯
(S)-1-[3,5-双(三氟甲基)苯基]-3-[1-(二甲基氨基)-3-甲基丁烷-2-基]硫脲
(R)富马酸托特罗定
(R)-(-)-盐酸尼古地平
(R)-(-)-4,12-双(二苯基膦基)[2.2]对环芳烷(1,5环辛二烯)铑(I)四氟硼酸盐
(R)-(+)-7-双(3,5-二叔丁基苯基)膦基7''-[((6-甲基吡啶-2-基甲基)氨基]-2,2'',3,3''-四氢-1,1''-螺双茚满
(R)-(+)-7-双(3,5-二叔丁基苯基)膦基7''-[(4-叔丁基吡啶-2-基甲基)氨基]-2,2'',3,3''-四氢-1,1''-螺双茚满
(R)-(+)-7-双(3,5-二叔丁基苯基)膦基7''-[(3-甲基吡啶-2-基甲基)氨基]-2,2'',3,3''-四氢-1,1''-螺双茚满
(R)-(+)-4,7-双(3,5-二-叔丁基苯基)膦基-7“-[(吡啶-2-基甲基)氨基]-2,2”,3,3'-四氢1,1'-螺二茚满
(R)-3-(叔丁基)-4-(2,6-二苯氧基苯基)-2,3-二氢苯并[d][1,3]氧杂磷杂环戊烯
(R)-2-[((二苯基膦基)甲基]吡咯烷
(R)-1-[3,5-双(三氟甲基)苯基]-3-[1-(二甲基氨基)-3-甲基丁烷-2-基]硫脲
(N-(4-甲氧基苯基)-N-甲基-3-(1-哌啶基)丙-2-烯酰胺)
(5-溴-2-羟基苯基)-4-氯苯甲酮
(5-溴-2-氯苯基)(4-羟基苯基)甲酮
(5-氧代-3-苯基-2,5-二氢-1,2,3,4-oxatriazol-3-鎓)
(4S,5R)-4-甲基-5-苯基-1,2,3-氧代噻唑烷-2,2-二氧化物-3-羧酸叔丁酯
(4S,4''S)-2,2''-亚环戊基双[4,5-二氢-4-(苯甲基)恶唑]
(4-溴苯基)-[2-氟-4-[6-[甲基(丙-2-烯基)氨基]己氧基]苯基]甲酮
(4-丁氧基苯甲基)三苯基溴化磷
(3aR,8aR)-(-)-4,4,8,8-四(3,5-二甲基苯基)四氢-2,2-二甲基-6-苯基-1,3-二氧戊环[4,5-e]二恶唑磷
(3aR,6aS)-5-氧代六氢环戊基[c]吡咯-2(1H)-羧酸酯
(2Z)-3-[[(4-氯苯基)氨基]-2-氰基丙烯酸乙酯
(2S,3S,5S)-5-(叔丁氧基甲酰氨基)-2-(N-5-噻唑基-甲氧羰基)氨基-1,6-二苯基-3-羟基己烷
(2S,2''S,3S,3''S)-3,3''-二叔丁基-4,4''-双(2,6-二甲氧基苯基)-2,2'',3,3''-四氢-2,2''-联苯并[d][1,3]氧杂磷杂戊环
(2S)-(-)-2-{[[[[3,5-双(氟代甲基)苯基]氨基]硫代甲基]氨基}-N-(二苯基甲基)-N,3,3-三甲基丁酰胺
(2S)-2-[[[[[((1S,2S)-2-氨基环己基]氨基]硫代甲基]氨基]-N-(二苯甲基)-N,3,3-三甲基丁酰胺
(2S)-2-[[[[[[((1R,2R)-2-氨基环己基]氨基]硫代甲基]氨基]-N-(二苯甲基)-N,3,3-三甲基丁酰胺
(2-硝基苯基)磷酸三酰胺
(2,6-二氯苯基)乙酰氯
(2,3-二甲氧基-5-甲基苯基)硼酸
(1S,2S,3S,5S)-5-叠氮基-3-(苯基甲氧基)-2-[(苯基甲氧基)甲基]环戊醇
(1S,2S,3R,5R)-2-(苄氧基)甲基-6-氧杂双环[3.1.0]己-3-醇
(1-(4-氟苯基)环丙基)甲胺盐酸盐
(1-(3-溴苯基)环丁基)甲胺盐酸盐
(1-(2-氯苯基)环丁基)甲胺盐酸盐
(1-(2-氟苯基)环丙基)甲胺盐酸盐
(1-(2,6-二氟苯基)环丙基)甲胺盐酸盐
(-)-去甲基西布曲明
龙蒿油
龙胆酸钠
龙胆酸叔丁酯
龙胆酸
龙胆紫-d6
龙胆紫

联系我们



关注我们

公众号

