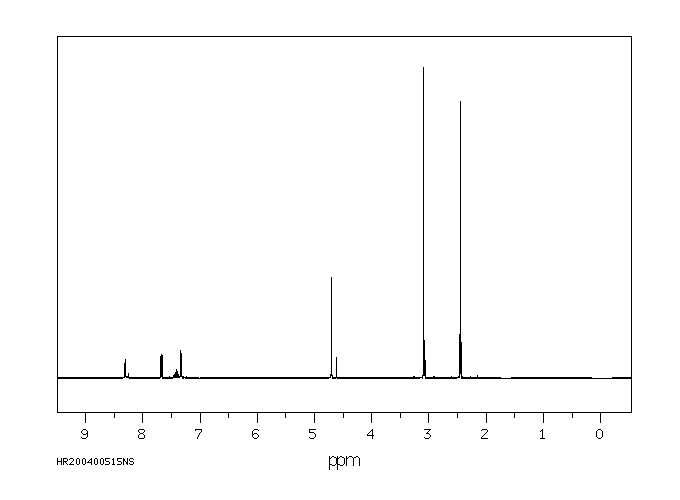
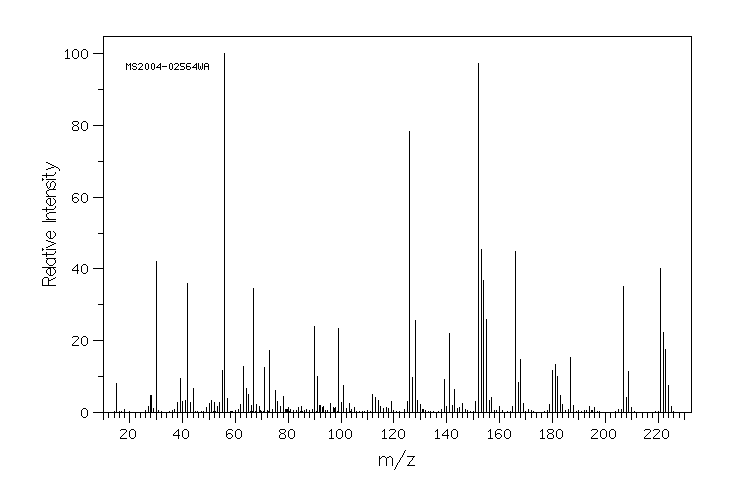
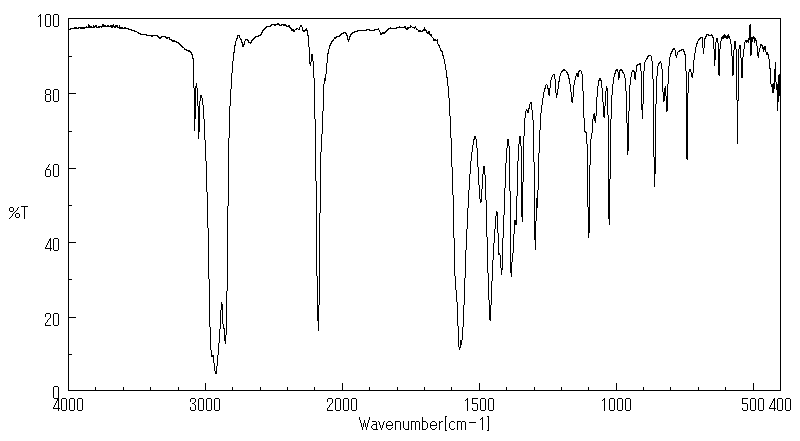
The in vivo metabolism of acetamiprid was studied in the honeybee, Apis mellifera L. The distribution of acetamiprid and its metabolites was monitored over a 72-h period in six biological compartments: head, thorax, abdomen, haemolymph, midgut and rectum. Honeybees were treated orally with 100 ug [(14)C]-acetamiprid/kg bee, a dose which is about 1500 times lower than the median lethal dose. After 72 hr, only 40% of the total radioactivity was eliminated, suggesting that acetamiprid and its metabolites tended to persist in the honeybee. Acetamiprid was rapidly distributed in all compartments and metabolized. Just after administration, radioactivity was mainly localized in the abdomen and subsequently in the rectum. After 72 hr, the maximum amount of radioactivity (about 20% of the ingested dose) was detected again in the abdomen, whereas the lowest level of total radioactivity was detected in the hemolymph. Radioactivity in the head did not exceed 7.6% of total ingested radioactivity. More than 50% of acetamiprid was metabolized in less than 30 min, indicating a very short half-life for the compound. During the first hours, acetamiprid was mainly detected in nicotinic acetylcholine receptor-rich tissues: abdomen, thorax and head. Of the seven metabolites detected, the major ones were 6-choronicotinic acid and an unknown metabolite called U1, which was present mainly in the rectum, the thorax and the head. Our results indicate that the low toxicity of acetamiprid may reflect its rapid metabolism.
BACKGROUND: Neonicotinoids, which are novel pesticides, have entered into usage around the world because they are selectively toxic to arthropods and relatively non-toxic to vertebrates. It has been suggested that several neonicotinoids cause neurodevelopmental toxicity in mammals. The aim was to establish the relationship between oral intake and urinary excretion of neonicotinoids by humans to facilitate biological monitoring, and to estimate dietary neonicotinoid intakes by Japanese adults. METHODOLOGY/PRINCIPAL FINDINGS: Deuterium-labeled neonicotinoid (acetamiprid, clothianidin, dinotefuran, and imidacloprid) microdoses were orally ingested by nine healthy adults, and 24 hr pooled urine samples were collected for 4 consecutive days after dosing. The excretion kinetics were modeled using one- and two-compartment models, then validated in a non-deuterium-labeled neonicotinoid microdose study involving 12 healthy adults. Increased urinary concentrations of labeled neonicotinoids were observed after dosing. Clothianidin was recovered unchanged within 3 days, and most dinotefuran was recovered unchanged within 1 day. Around 10% of the imidacloprid dose was excreted unchanged. Most of the acetamiprid was metabolized to desmethyl-acetamiprid. Spot urine samples from 373 Japanese adults were analyzed for neonicotinoids, and daily intakes were estimated. The estimated average daily intake of these neonicotinoids was 0.53-3.66 ug/day. The highest intake of any of the neonicotinoids in the study population was 64.5 ug/day for dinotefuran, and this was <1% of the acceptable daily intake.
... Five males and five female /rats/ were orally administered a daily dose of non-labelled acetamiprid for 14 days followed by a single dose of radiolabelled acetamiprid on day 15. The urine and feces were collected once on day 14 and then at 24-hour intervals after administration of the [(14)C]acetamiprid dose solution until sacrifice. Qualitative analysis of metabolites was performed by thin-layer co-chromatography with unlabelled reference substances. The unknown metabolite was identified by liquid chromatography-tandem mass spectrometry (LC-MS/MS) as the glycine conjugate of IC-O (abbreviated as IC-O-Gly). The major radioactive compounds in the excreta of rats were acetamiprid itself (males: 5.21%; females: 7.41%), demethylated compound IM-2-1 (males: 15.48%; females: 20.39%), nicotinic acid derivative IC-O (males: 11.12%; females: 8.01%) and IC-O glycine conjugate IC-O-Gly (males: 10.10%; females: 10.32%). In addition, MeS-IC-O, IM-1-4, IM-2-4, IM-O, IM-1-3 and IM-2-3 were detected, but they accounted for less than 2% of the dose. There were several unknown compounds in urine, and the maximum abundance of an unknown compound in the "others" fraction was 1.0%. It was considered that the major metabolic routes of acetamiprid in rats are the production of IM-2-1 by N-demethylation, the production of IC-O by detachment of the cyanoacetamide side-chain from IM-2-1, and the production of IS-1-1 and IS-2-1 by detachment of the cyanoacetamide sidechain from acetamide and IM-2-1, respectively.
... Radioactive compounds in the excreta of rats were identified and analysed quantitatively. The major compounds identified were acetamiprid itself (males: 6.10%; females: 5.63%), demethylated compound IM-2-1 (males: 19.51%; females: 19.00%) and nicotinic acid derivative IC-O (males: 28.19%; females: 25.52%) in group B; acetamiprid (males: 7.75%; females: 7.34%), IM-2-1 (males: 24.48%; females: 21.37%) and IC-O (males: 27.11%; females: 27.63%) in group D; and acetamiprid (males: 4.16%; females: 6.12%), IM-2-l (males: 13.39%; females: 18.98%) and IC-O (males: 28.13%; females: 24.73%) in group A. Acetamiprid (males: 3.98%; females: 4.51%), IM-2-1 (males: 16.95%; females: 16.56%), IS-1-1 (males: 13.15%; females: 16.45%) and IS-2-1 (males: 35.61%; females: 30.23%) were detected as the main compounds in group CN-B. IS-1-1 and IS-2-1 were thought to be generated by cleavage of the side-chains of acetamiprid and IM-2-1. In addition, IC-O-Gly, MeS-IC-O, IM-1-4, IM-2-4, IM-O, IM-1-3 and IM-2-3 were detected in groups A, B and D, but each at less than 4% of the dose. The main metabolic pathways of acetamiprid in rats were the transformation to IM-2-1 by demethylation and further to IC-O after cleaving IS-1-1 and IS-2-1 from acetamiprid and IM-2-1, respectively
Organic nitriles are converted into cyanide ions through the action of cytochrome P450 enzymes in the liver. Cyanide is rapidly absorbed and distributed throughout the body. Cyanide is mainly metabolized into thiocyanate by either rhodanese or 3-mercaptopyruvate sulfur transferase. Cyanide metabolites are excreted in the urine. (L96)
IDENTIFICATION AND USE: Acetamiprid is a solid. Acetamiprid is a neonicotinoid insecticide that is used for the control of sucking-type insects on leafy vegetables, fruiting vegetables, cole crops, citrus fruits, pome fruits, grapes, cotton, and ornamental plants and flowers. HUMAN STUDIES: There are two cases of acute poisoning with an insecticide formulation containing acetamiprid for suicidal purposes. Both cases experienced severe nausea and vomiting, muscle weakness, hypothermia, convulsions, and clinical manifestations including tachycardia, hypotension, electrocardiogram changes, hypoxia, and thirst in the case with the higher serum concentration of acetamiprid. The symptoms were partially similar to acute organophosphate intoxication. A 79-year-old man had ingested acetamiprid and got medical attention two hours after ingestion. On arrival, his blood concentration of acetamiprid was 21.1 ug/mL. He had consciousness disturbance, hypotension, nausea, vomiting and hyperglycemia, but had no constricted pupils nor mucous supersecretion which are characteristic in organophosphate poisoning. In human peripheral blood lymphocytes, in vitro acetamiprid induced sister chromatid exchanges and chromosomal aberrations significantly at all concentrations and treatment times and micronucleus formation was significantly induced at 30, 35, and 40 ug/mL of acetamiprid. ANIMAL STUDIES: Acetamiprid was not an irritant in a study of ocular and dermal irritation in rabbits or a dermal sensitizer in the Magnusson and Kligman maximization test in guinea-pigs. Acetamiprid was admixed to the diet of mice at dose levels of 0, 400, 800, 1600, or 3200 ppm and fed to mice for a period of 13 weeks. 2 males and 2 females at 3200 ppm died during the study. Tremor was observed in 5 females at 3200 ppm. A treated-related increase in mean relative liver weight was observed in both sexes at 800, 1600, and 3200 ppm. Microscopic examination revealed centrilobular hepatocellular hypertrophy and depletion of fat from the adrenal cortex in both sexes at 3200 ppm. Developmental toxicity noted in rats (NOEL = 16 mg/kg/day, based on the skeletal variation) and rabbits (developmental NOEL = 15 mg/kg/day, based on fused thoracic vertebral arches and fused ribs in 2 fetuses). In rats, acetamiprid disrupted testosterone biosynthesis by decreasing the rate of conversion of cholesterol to testosterone and by preventing cholesterol from entering the mitochondria within the Leydig cells. These effects caused reproductive damage to the rats. Acetamiprid was neurotoxic in rats and induced neurodevelopmental toxicity in mice. Acetamiprid exposure interfered with the development of the neural circuits required for executing socio-sexual and anxiety-related behaviors in male mice. Acetamiprid was negative when Salmonella typhimurium strains TA100, TA1535, TA98, and TA1537, and Escherichia coli strain WP2 uvrA were used in reverse mutation tests with and without metabolic activation. ECOTOXICITY STUDIES: Acetamiprid can cause neuropathic changes in sciatic nerve of frogs. After oral consumption acetamiprid increased sensitivity of honeybees to antennal stimulation by sucrose solutions at doses of 1 ug/bee and impaired long-term retention of olfactory learning at the dose of 0.1 ug/bee. Acetamiprid thoracic application induced no effect in these behavioral assays but increased locomotor activity (0.1 and 0.5 ug/bee) and water-induced proboscis extension reflex (0.1, 0.5, and 1 ug/bee). Acetamiprid had significant adverse effects on different development life stages of Amblyseius cucumeri. Acetamiprid exposure caused oxidative stress and DNA damage of earthworm and changed the activity of the antioxidant enzyme.
Organic nitriles decompose into cyanide ions both in vivo and in vitro. Consequently the primary mechanism of toxicity for organic nitriles is their production of toxic cyanide ions or hydrogen cyanide. Cyanide is an inhibitor of cytochrome c oxidase in the fourth complex of the electron transport chain (found in the membrane of the mitochondria of eukaryotic cells). It complexes with the ferric iron atom in this enzyme. The binding of cyanide to this cytochrome prevents transport of electrons from cytochrome c oxidase to oxygen. As a result, the electron transport chain is disrupted and the cell can no longer aerobically produce ATP for energy. Tissues that mainly depend on aerobic respiration, such as the central nervous system and the heart, are particularly affected. Cyanide is also known produce some of its toxic effects by binding to catalase, glutathione peroxidase, methemoglobin, hydroxocobalamin, phosphatase, tyrosinase, ascorbic acid oxidase, xanthine oxidase, succinic dehydrogenase, and Cu/Zn superoxide dismutase. Cyanide binds to the ferric ion of methemoglobin to form inactive cyanmethemoglobin. (L97)
来源:Toxin and Toxin Target Database (T3DB)
毒理性
致癌物分类
对人类无致癌性(未列入国际癌症研究机构IARC清单)。
No indication of carcinogenicity to humans (not listed by IARC).
来源:Toxin and Toxin Target Database (T3DB)
毒理性
副作用
神经毒素 - 其他中枢神经系统神经毒素
Neurotoxin - Other CNS neurotoxin
来源:Haz-Map, Information on Hazardous Chemicals and Occupational Diseases
毒理性
毒性数据
LC50 (rat) = 290 mg/m3
大白鼠 LC50 = 290 毫克/立方米
LC50 (rat) = 290 mg/m3
来源:Haz-Map, Information on Hazardous Chemicals and Occupational Diseases
To obtain information on the absorption, distribution, rate and route of elimination, metabolism and pharmacokinetics of acetamiprid, a study was performed in adult Sprague-Dawley rats (body weight 154-193 g for males, 134-152 g for females; aged 5-6 weeks at the start of dosing; dosing for 15 days) using [(14)C]acetamiprid. The radiolabelled test substance (chemical purity > 99.9%, radiochemical purity 97.1-97.2%) was sent by the sponsor to the contract research organization. The non-labelled test substance /had/ a chemical purity of greater than 99.9%. The studies were conducted after oral administration of the test substance for 15 days. In total, five treatment groups (groups I, II, III, IV and V), consisting of 6 rats (3 males and 3 females) in each of the first three groups and 10 rats (5 males and 5 females) in each of the two remaining groups, were used. A single control group (group VI), consisting of four rats (two males and two females), was used. Groups I, II and III received oral doses of [(14)C]acetamiprid in 0.9% saline for 15 days at a target dose rate of 1.0 mg/kg body weight (bw). Groups IV and V received oral doses of acetamiprid in 0.9% saline for 14 days followed by a single oral dose of [(14)C]acetamiprid in 0.9% saline on day 15. The actual dose rate was 0.97-1.01 mg/kg bw for the rats in all five groups. The radiochemical purity of [(14)C]acetamiprid in the dose solution was determined to be 97.9% by high-performance liquid chromatographic (HPLC) analysis. The dose solution was stable under refrigerated conditions for at least 15 days. The specific activity of the radiolabelled dose solution was determined to be 1.85 x 10(+3) Bq/ug. Group VI was dosed with 0.9% saline only. Rats of groups I, II and III were sacrificed 1, 10 and 96 hours, respectively, after dosing of [(14)C]acetamiprid for 15 days. Rats of group IV were sacrificed 96 hours after a single dose of [(14)C]acetamiprid for tissue and organ collection. Group V was used only for blood pharmacokinetic analysis. Whole blood was drawn from each rat of group III approximately 1 hour post-dosing on days 1, 3, 7 and 15 to determine the [(14)C]acetamiprid concentration in blood. The average concentration in blood was in the range of 0.477-0.747 ug/mL in the males and 0.465-0.698 ug/mL in the females. Variation between animals was observed. These results indicate that the blood concentration at 1 hour post-dosing was consistent during the entire dosing period. Whole blood was drawn from each rat of group V at approximately 0.25, 0.5, 1, 2, 3, 4, 5, 7, 9, 12, 24 and 48 hours to determine the [(14)C]acetamiprid concentration in blood. The mean values for peak concentration (Cmax), time to Cmax (Tmax), absorption half-life and area under the concentration versus time curve at infinity for the male rats were 0.798 +/- 0.111 ug/mL, 2.80 +/- 0.637 hours, 1.35 +/- 0.825 hours and 8.35 +/- 1.12 ug eq*hr/mL, respectively. Values for the same parameters in female rats averaged 0.861 +/- 0.132 ug/ml, 2.81 +/- 0.894 hours, 1.18 +/- 0.868 hours and 10.3 +/- 2.90 ug eq*hr/mL, respectively. The elimination half-lives for the male and female rats were 4.42 +/- 1.10 hours and 5.56 +/- 1.93 hours, respectively. The pharmacokinetic parameters for both sexes did not differ considerably. The Tmax values in both sexes indicated that the rate of absorption of acetamiprid was rapid, and a maximum blood concentration to possible saturation was achieved in approximately 2-3 hours. The elimination results indicate that most acetamiprid (53-65%) was excreted in the urine. The excretion in urine and cage rinse combined amounted to 61-73%. The results also indicate that acetamiprid was absorbed rapidly (within 1 hour) from the gastrointestinal tract, as greater than 90% of the administered dose was eliminated from the gastrointestinal tract within 1 hour after dosing. No difference was observed in elimination of test substance between chronic administration of acetamiprid for 14 days followed by a single administration of radiolabelled acetamiprid on day 15 (group IV) and chronic administration of radiolabelled acetamiprid for 15 days (groups I, II and III). The amount of administered radioactivity eliminated in feces was lower for females (22-29%) than for males (30-35%). The whole blood, liver, kidney, lung, pancreas, spleen, heart, brain, testes (male), ovary (female), skeletal muscles, inguinal fat (white), skin with hair, thyroid, bone, adrenal glands, gastrointestinal tract with contents, cage rinses and residual carcasses were collected from each rat of groups I, II, III and IV. All collected samples were not composited but kept and analysed separately to account for the material balance for each rat. Radioactivity, after administration of the last chronic dose, was detected at the earliest sampling point (1 hour) in all the tissues collected from each rat. The radioactivity in most tissues was the highest at 1 hour post-dosing and declined rapidly thereafter (groups II and III). The Tmax for [(14)C]acetamiprid in the male and female rats indicated that the rate of absorption was rapid, and a maximum blood concentration (approximately 0.8 ug/mL) to possible saturation was achieved in approximately 2-3 hours. The levels of [(14)C]acetamiprid residue in tissues collected at 1 hour post-dosing confi rm the results obtained from the pharmacokinetic analysis. [(14)C]Acetamiprid residue levels seen in tissues collected 10 hours post-dosing (group II) were found to be substantially lower than residue levels in tissues collected 1 hour post-dosing. The elimination half-life for both sexes indicated that the rate of elimination was rapid. The levels of [(14)C]acetamiprid residues in tissues collected at 10 hours post-dosing confirm the results obtained from the pharmacokinetic studies. [(14)C]Acetamiprid residue levels seen in tissues collected 96 hours post-dosing (group III) were found to be very low compared with the levels observed in the tissues collected at 1 hour and 10 hours post-dosing. The elimination half-life for both sexes was between 4 and 6 hours postdosing, indicating that the rate of elimination was rapid and that retention of residue in tissues after chronic administration was minimal. The highest radioactivity levels were observed in the gastrointestinal tract, liver and kidney in both sexes at all sacrifice times. The lowest concentration was observed in bone and white fat. The residue levels observed were higher in all tissues of rats chronically treated with [(14)C]acetamiprid for 15 days (group III) compared with the rats in group IV, which received a single final dose of [(14)C]acetamiprid following 14 days of non-labelled acetamiprid doses. The residue levels observed in the tissues of rats sacrificed 96 hours after the last dose were very low (0.01-0.1 part per million [ppm]), as most of the administered dose (> 90%) was eliminated through the urine and feces. The total administered radioactivity recovered in groups I, II, III and IV was in the range of 91.7-106%, whereas recovery in group V (the pharmacokinetics group) was 71.7% and 85.6% in males and females, respectively. The loss of urine samples during a series of bleeding procedures is a possible explanation for the low recovery in group V.
To ascertain the effect of administration of acetamiprid in single low and high doses, the absorption, distribution, metabolism and excretion of acetamiprid in rats were investigated. [Pyridine- 2,6-(14)C]acetamiprid was intravenously or orally administered to five male and five female rats in groups A, B and D at dose levels of 1.0, 1.0 and 50 mg/kg bw, respectively. In group CN-B, the metabolism study of [cyano-(14)C]acetamiprid was performed at a dose level of 1.0 mg/kg bw. Group A was for the determination of the absorption rate by calculation from the excretion rate and metabolite analysis. Groups B, D and CN-B were for blood levels, tissue distribution, metabolite analysis and excretion rate. ... In summary, acetamiprid orally dosed in rats was rapidly absorbed and widely distributed into the tissues via blood. The majority of the radioactivity was excreted in the urine through the kidney and in the feces via bile. The disappearance of the radioactivity from the body of the rat was rapid, and there were no tissues that are presumed to accumulate the compound. No differences in the sexes were observed.
A biliary excretion study was conducted using Sprague-Dawley bile duct-cannulated rats approximately 10-12 weeks old at dosing. Four male and four female bile duct-cannulated rats received single doses of [(14)C]acetamiprid in 0.9% saline through an intragastric cannula. The average dose rates were 1.02 and 1.07 mg/kg bw for the male and female rats, respectively. The radiochemical purity of [(14)C]acetamiprid in the dose solution was determined to be 97.1% by HPLC analysis. One male and one female rat were dosed with placebo (0.9% saline, containing no test substance). A steady increase in [(14)C]acetamiprid residue level was observed in bile from 3 to 12 hours post-dosing, with the highest amount (percentage of administered dose) at 12 hours post-dosing in both male and female rats. The average recovery of the administered dose in bile over a 48-hour period was 19.9% +/- 1.47% in the male rats and 18.6% +/- 0.62% in the female rats. Recovery of the [(14)C]acetamiprid residues excreted in bile accounted for less than 20% of the total administered dose, suggesting that bile is not a predominant excretory pathway in either the male or the female rats. The absorption of the test substance and the extent of fi rst-pass metabolism/presystemic elimination were not signifi cantly different between the sexes. were not significantly different between the sexes. The average recovery of the administered dose in feces over a 48-hour period was 6.72% 3.36% in the male rats and 5.84% +/- 0.86% in the female rats. The average recovery of the administered dose in urine over a 48-hour period was 24.3% +/- 5.22% in the male rats and 36.9% +/- 3.80% in the female rats. In the male and female rats, the sum of urine plus cage rinses, 60.2% +/- 5.20% and 64.4% +/- 2.86%, respectively, accounted for the major residues, suggesting that most of the administered dose was excreted in urine. The average recovery of the administered dose in liver at 48 hours post-dosing was 0.22% 0.13% in the male rats and 0.18% +/- 0.18% in the female rats. The average recovery of the administered dose in the gastrointestinal tract at 48 hours post-dosing was 0.46% +/- 0.34% in the male rats and 0.33% +/- 0.23% in the female rats. These results indicate that an insignificant amount of acetamiprid (< 1% in the collected tissues) was absorbed into the liver or remained in the gastrointestinal tract in both the male and female rats. the male and female rats. The total recoveries of the administered dose in the three male rats were 93.2%, 92.8% and 89.6%, respectively. The total recoveries of the administered dose in the three female rats were 94.9%, 93.5% and 91.2%, respectively.
The extent of absorption of acetamiprid was studied following application of 70% wettable powder containing [(14)C]acetamiprid (purity 97.5%) to the skin of male Crl: CD(SD)BR rats. The animals were approximately 8 weeks old upon arrival and weighed 176-216 g (preliminary phase) and 143-203 g (defi nitive phase). Target dose levels were 1, 10 and 100 ug/sq cm. Actual dose levels were 0.0136 mg/animal (1.09 ug/sq cm), 0.119 mg/animal (9.53 ug/sq cm) and 1.13 mg/animal (90.2 ug/sq cm). A preliminary phase, consisting of two groups of four animals each, was conducted to evaluate and establish test material application and skin washing techniques. In the preliminary phase, male rats were dermally dosed at two levels (0.0128 mg/animal and 1.26 mg/animal). In the definitive phase, three groups of 24 rats per group were dermally dosed with [(14)C]-acetamiprid at three dose levels. A control group of two rats received only the vehicle (1% carboxymethylcellulose aqueous solution). Urine and feces were collected from each rat. Immediately before sacrifice, the skin at the application site was washed. Four rats per time point from each dose group were sacrificed at 0.5, 1, 2, 4, 10 and 24 hours; the control rats were sacrifi ced at 24 hours. At sacrifice, blood was collected by cardiac puncture. Among the treated groups, the mean total recovery of radioactivity ranged from 96.6% to 102%, with most of the radioactivity (63.9-87.5%) in the skin wash. Radioactivity in the skin at the application site accounted for 10.2-32.2% of the applied radioactivity. Radioactivity in blood, excreta and carcasses accounted for less than 6.50% of the applied radioactivity. The amounts of radioactivity found in the blood, eliminated in the excreta and retained in the carcass were considered to result from direct dermal absorption of [(14)C]acetamiprid. Within groups, amounts of dermal absorption increased with increasing exposure time. The highest absorption was detected at the longest exposure time, 24 hours post-dosing, and accounted for 4.27% (0.581 ug), 6.34% (7.54 ug) and 2.82% (31.9 ug) for the 1.09, 9.53 and 90.2 ug/sq cm dose groups, respectively. The sum of direct absorption and amount of radioactivity remaining in the skin at the application site was considered to be indirect absorption. The amounts of indirect absorption were 3-5 ug, 25-37 ug and 118-197 ug for the 1.09, 9.53 and 90.2 ug/sq cm dose groups, respectively. The highest concentration of radioactivity in blood was 0.001 ppm for the 1.09 ug/sq cm dose group at 24 hours post-dosing, 0.019 ppm and 0.010 ppm for the 9.53 ug/sq cm dose group at 10 and 24 hours post-dosing, respectively, and 0.041 ppm for the 90.2 ug/sq cm dose group at 24 hours post-dosing. The amount of direct absorption of acetamiprid in rats was proportional at the two lower dose levels and appeared to reach saturation at the highest dose level.