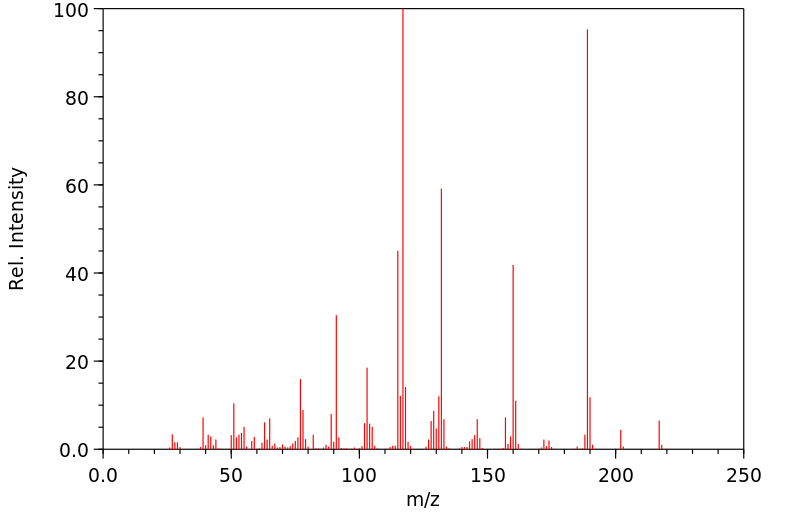
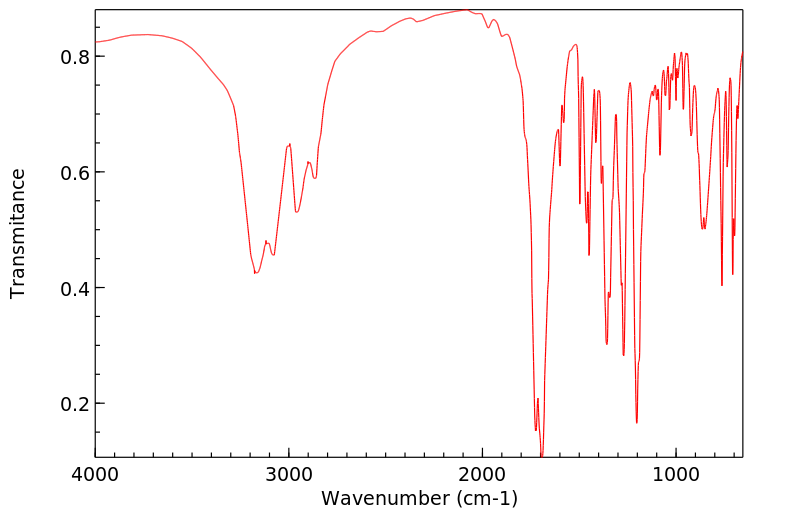
The metabolic fate of glutehimide has not been completely elucidated, particularly following toxic doses of the drug. There is considerable evidence that therapeutic doses of glutethimide are almost completely metabolized in the liver by hydroxylation of the ethyl side chain (levorotatory isomer) and excreted in urine chiefly as glucuronides. About 2% of a dose is metabolized to glutaconimide which has some hypnotic activity. ... 4-Hydroxy-2-ethyl-2 phenylglutarimide is a metabolite which accumulates in the plasma and tissues (including the brain) of patients who have ingested large doses of glutethimide. Several phenolic metabolites and others yet to be identified have been found in urine following acute glutethimide overdosage.
Although glutethimide blood concentrations greater than 10 ug/mL are generally associated with intoxication, there is a poor correlation between glutethimide plasma concentration and the clinical course of the patient, possibly because of the formation and accumulation of an active metabolite, 4-hyroxy-2-ethyl-2-phenylglutarimide.
Glutethimide has an asymmetric carbon atom and gives two optical isomers, both of which are metabolized in a different fashion. The D-isomer is hydrolyzed at the glutarimide ring, loses water, and breaks down into alpha-phenyl-alpha-ethylglutaconimide, which is excreted in the urine as a nonconjugated metabolite in approximately 2% of the administered dose. A major portion of the hydrolyzed D-isomer is combined with glucuronic acid and is excreted in the urine in approximately 45% of the administered dose. The L-isomer is hydrolyzed with release of acetaldehyde from a-phenyl glutarimide. This metabolite is isolated in the urine in approximately 4 percent of the administered dose. The remaining major portion is also combined with glucuronic acid and is excreted in the urine in approximately 45 % of the administered dose. Both glucuronides are water soluble but not fat soluble and no longer possess sedative activity.
IDENTIFICATION: Glutethimide is used as a hypnotic agent. Glutethimide is colorless or white, odorless crystals or powder. It is practically insoluble in water, soluble in ethanol, chloroform and ether. Freely soluble in acetone and ethyl acetate and soluble in methyl alcohol. Stability: at pH 5 the chemical half life was 28.3 years at 25 °C and 1.02 months at pH 8, the decomposition being due to hydrolysis. Indications: Used as an hypnotic in insomnia but rarely as a sedative, glutethimide was initially believed to be almost free from side effects. However, further experience of its toxicity and because its dependence liability, glutethimide has been banned in many countries and many companies have stopped production. HUMAN EXPOSURE: Main risks and target organs: The main target organ is the central nervous system causing coma with fluctuations in depth, and various degrees of hypotension. Anticholinergic effects often occur. Summary of clinical effects: At lower doses, acute intoxication may cause somnolence, ataxia, tonic muscle spasms and abnormal reflexes. In severe intoxication, hypotension, hypothermia, shock, coma, respiratory depression and acidosis may occur. Effects on other organs are usually secondary to coma and shock. Contraindications: Glutethimide is contraindicated in porphyria. Due to its anti-muscarinic action, it should be given with great care to patients with closed-angle glaucoma, prostatic hypertrophy or urinary tract obstruction, and certain cardiac arrhythmias. Alcohol enhances absorption and the hypnotic effects of glutethimide. Like barbiturates, glutethimide induces microsomal hepatic enzymes and enhances the metabolism of coumarin anticoagulants and other drugs, lowering their plasma concentrations. Chronic administration of glutethimide may also enhance vitamin D metabolism. Absorption by route of exposure: Oral: In six healthy volunteers given a dose of 500 mg, absorption was irregular and peak plasma concentrations occurred over one to six hours. However, in four of the six subjects absorption was biphasic. Erratic absorption may be due to the poor solubility of glutethimide in water. Distribution by route of exposure: Oral: following oral administration of 500 mg of glutethimide to healthy subjects, peak plasma concentrations were achieved within two to six hours. Plasma protein binding of glutethimide was about 50%. Glutethimide concentrations were found in the breast milk of 13 nursing mothers given glutethimide. Glutethimide is highly lipophilic and rapidly concentrates in brain and adipose tissue. Biological half-life by route of exposure: Oral: The elimination half-life is 10 to 12 hours, but may increase in severe poisoning. In six healthy subjects, initial half-lives after ingestion were 2.7 to 4.3 hours and subsequent half-lives ranged from 5.1 to 22 hours. Metabolism: Glutethimide is partially metabolized by hydroxylation into 4-hydroxy-2-ethyl-2-phenylglutarimide. In the mouse, this appears to be twice as potent as the parent compound in mice and is believed to contribute to prolonged coma following overdosage. Hydroxylated metabolites are conjugated and excreted mainly in the urine but also in bile. Elimination by route of exposure: Oral: glutethimide is inactivated by conjugation and the metabolites are excreted in urine, only 2% of the parent substance is excreted in urine, up to 2% of the dose has been reported to be found in the feces. Mode of action: Toxicodynamics Pharmacodynamics: Glutethimide directly blocks electron transfer in cellular respiration. Teratogenicity: There is no evidence of teratogenicity from therapeutic use. Interactions: The effects of glutethimide are additive with those of benzodiazepines, barbiturates, codeine and other CNS depressants. Concomitant administration of antidepressants, antiparkinsonian drugs or other anticholinergic agents may cause additive anticholinergic effects such as urinary retention, exacerbation of glaucoma, or adynamic ileus. Ethanol enhances the effects of glutethimide. Glutethimide induces the hepatic metabolism of some drugs, such as dicoumarol derivatives, the dose of drugs taken concomitantly may require adjustment. Main adverse effects: Common adverse effects are as follows: nausea, headache, hangover, blurred vision, occasional skin rashes, blood disorders (megaloblastic anemia). Osteomalacia peripheral neuropathy and cerebral impairment after prolonged use may also occur. Glutethimide is a drug of abuse and may cause dependence. Acute poisoning: Ingestion: Ingestion is the only route by which acute poisoning may occur. Mild intoxication with small doses results in somnolence, ataxia, tonic muscle spasms, abnormal reflexes. In severe intoxication coma, hypotension, hypothermia, shock, respiratory depression and cerebral edema may occur. Signs from other organs and systems are usually secondary to coma and shock. Chronic poisoning: Ingestion: Ingestion is the only route of glutethimide administration in humans. Prolonged use of the drug may cause peripheral neuropathy, hypocalcemia. Acute abstinence syndrome following glutethimide withdrawal has been described. Chronic ingestion of high doses is associated with impaired memory, inability to concentrate, ataxia, tremors, hyporeflexia, slurring of speech and convulsions. Course, prognosis, cause of death: Any acute poisoning without loss of consciousness may be regarded as mild and the patient is not at risk. The occurrence of coma, hypotension, hypothermia, shock, respiratory depression and complications such as pneumonia mean that glutethimide poisoning is potentially serious. However, death is unusual provided intensive care when needed. Cerebral edema may be fatal. Systematic description of clinical effects Cardiovascular: Hypotension, shock and tachycardia have been observed. Unexplained dysrhythmias may be due to the antimuscarinic effects of the drug or low plasma calcium concentrations. Respiratory: Respiratory depression with intermittent apnea and or arrest may occur in very severe cases. Pneumonia due to aspiration and pulmonary oedema have been reported. Neurological: Central Nervous System (CNS): Various degrees of CNS depression may occur, ranging from lethargy to deep coma. Cerebral edema, intracranial hemorrhage, tonic muscle spasms and hyperreflexia may occur. Truncal ataxia has been reported in acute glutethimide intoxication in children. Peripheral nervous system: Peripheral neuropathy and diplopia has been reported following chronic use. Autonomic nervous system: Glutethimide has antimuscarinic/anticholinergic activity, tachycardia, dryness of mouth, mydriasis, irritability, urinary retention and constipation. Skeletal and smooth muscle: Tonic muscle spasm and paralytic ileus (adynamic ileus) may also be observed. Gastrointestinal: Gastrointestinal atony due to parasympatholytic activity may occur. Urinary: Renal: With the exception of possible pre-renal uremia due to severe hypotension, no other renal effects occur. Other: Urinary retention may occur due to the anticholinergic effect of glutethimide. Dermatological: Bullous changes resembling those seen in barbiturate poisoning and erythematous vesicles have been described. Eye, ear, nose, throat: local effects: Mydriasis and papilloedema have been observed. Hematological: Significant methemoglobinemia has been reported rarely. In a further case, megaloblastic anemia, thrombocytopenia and aplastic anemia occurred. Metabolic: Acid-base disturbances: Acid base disturbances may occur secondary to coma or shock. Fluid and electrolyte disturbances: Hypocalcaemia has been described. Others: Hypothermia has been described. Special risks: Glutethimide readily crosses the placenta and may cause neonatal respiratory depression and neonatal withdrawal symptoms. Eight to 12 hours after a maternal dose of glutethimide. It was found in breast milk. ANIMAL/PLANT STUDIES: Parenteral: following intraperitoneal administration of glutethimide in rats, most of drug was found in the brain and spinal cord and other fat-containing tissues after 20 minutes.
A series of glutethimide congeners produce concentration-dependent inhibition of corticosterone production by a suspension of isolated rat adrenal cells. The dextro-rotatory antipode of aminoglutethimide is more potent than its levo enantiomer in inhibiting corticosterone production in this system. Glutethimide, its metabolite glutaconimide, and congeners including those with anti-convulsant activity, 4-hydroxyglutaconimide and 4-aminoglutethimide, have all demonstrated concentration-dependent inhibition of corticosterone production by isolated rat adrenal cells.
The intensity, uniformity and time course of anticoagulant interference by phenobarbital, secobarbital, glutethimide, chloral hydrate and methaqualone were systematically investigated in 16 patients receiving coumarin therapy. Each subject received an individualized fixed daily dose of warfarin and served as his own pre- and postsedative treatment control. Prothrombin times were measured four times weekly during five long-term experiments. Anticoagulant inhibition was observed during the administration of phenobarbital, secobarbital and glutethimide; there was no significant change in prothrombin test results during the trials of chloral hydrate and methaqualone. Barbiturates and glutethimide should not be administered to patients receiving coumarin drugs. The concurrent use of drugs from these groups is decreasing according to a survey of 200 hospital medical records. Chloral hydrate and methaqualone interact pharmacologically with orally administered anticoagulant agents, but the effect is not clinically significant. It is concluded that chloral hydrate and methaqualone may be administered safely without additional caution in prothrombin test monitoring during oral anticoagulant therapy.
Glutethimide, like barbiturates, may induce hepatic microsomal enzymes resulting in increased metabolism of coumarin anticoagulants and decreased anticoagulant response. If a hypnotic is required in patients receiving oral anticoagulants, it is preferable to use an alternative hypnotic (e.g., a benzodiazepine) which does not alter the anticoagulant response. Patients maintained on both glutethimide and a coumarin anticoagulant have a risk of hemorrhage if glutethimide is discontinued and the dosage of the anticoagulant is not adjusted. Glutethimide therapy should not be initiated or discontinued in patients receiving oral anticoagulants without careful attention to the possible need for adjusting anticoagulant dosage.
Concomitant administration of glutethimide and tricyclic antidepressants such as amitriptyline or imipramine may produce additive anticholinergic effects. Although additive anticholinergic effects are usually minor, the possibility of precipitating adynamic ileus, urinary retention, or acute glaucoma, especially in elderly patients, should be considered.
glutethimide is inactivated by conjugation and the metabolites are excreted in urine, only 2% of the parent substance is excreted in urine, up to 2% of the dose has been reported to be found in the faeces.
来源:DrugBank
吸收、分配和排泄
Glutethimide appears to be irregularly absorbed from the GI tract. Plasma concentrations of the drug required for sedative or hypnotic effects are not known. In limited studies, oral administration of 500 mg and 1 g single doses of glutethimide produced peak plasma concentrations of 2.9-7.1 ug/mL and 6.2-6.8 ug/mL, respectively, within 1-6 hours. The onset of action of glutethimide is rapid; sleep is usually induced within 30 minutes and lasts 4-8 hours following usual hypnotic doses.
Glutethimide appears to be irregularly absorbed from the GI tract. Plasma concentrations of the drug required for sedative or hypnotic effects are not known. In limited studies, oral administration of 500 mg and 1 g single doses of glutethimide produced peak plasma concentrations of 2.9-7.1 ug/mL and 6.2-6.8 ug/mL, respectively, within 1-6 hours. The onset of action of glutethimide is rapid; sleep is usually induced within 30 minutes and lasts 4-8 hours following usual hypnotic doses.
Once absorbed, glutethimide quickly becomes concentrated in organs containing fat such as brain and adipose tissues, with a volume of distribution larger than that of whole body water. The drug recirculates from fat stores and is carried to the liver where it undergoes biotransformation, somewhat reminiscent of redistribution of ultra short acting barbiturates like thiopental.
Distribution studies indicate that there is extensive tissue localization of glutethimide, particularly in adipose tissue. In addition, glutethimide (and/or its metabolites) has been detected in liver, kidneys, brain, and bile. Glutethimide crosses the placenta, and small quantities of the drug are distributed into milk. When 1 g doses of glutethimide were administered to pregnant women 2 hours before delivery, maternal and neonatal plasma concentrations of glutethimide were essentially the same immediately after delivery.
[EN] COMPOUNDS AND THEIR USE AS BACE INHIBITORS<br/>[FR] COMPOSÉS ET LEUR UTILISATION EN TANT QU'INHIBITEURS DE BACE
申请人:ASTRAZENECA AB
公开号:WO2016055858A1
公开(公告)日:2016-04-14
The present application relates to compounds of formula (I), (la), or (lb) and their pharmaceutical compositions/preparations. This application further relates to methods of treating or preventing Αβ-related pathologies such as Down's syndrome, β- amyloid angiopathy such as but not limited to cerebral amyloid angiopathy or hereditary cerebral hemorrhage, disorders associated with cognitive impairment such as but not limited to MCI ("mild cognitive impairment"), Alzheimer's disease, memory loss, attention deficit symptoms associated with Alzheimer's disease, neurodegeneration associated with diseases such as Alzheimer's disease or dementia, including dementia of mixed vascular and degenerative origin, pre-senile dementia, senile dementia and dementia associated with Parkinson's disease.
[EN] METHYL OXAZOLE OREXIN RECEPTOR ANTAGONISTS<br/>[FR] MÉTHYLOXAZOLES ANTAGONISTES DU RÉCEPTEUR DE L'OREXINE
申请人:MERCK SHARP & DOHME
公开号:WO2016089721A1
公开(公告)日:2016-06-09
The present invention is directed to methyl oxazole compounds which are antagonists of orexin receptors. The present invention is also directed to uses of the compounds described herein in the potential treatment or prevention of neurological and psychiatric disorders and diseases in which orexin receptors are involved. The present invention is also directed to compositions comprising these compounds. The present invention is also directed to uses of these compositions in the potential prevention or treatment of such diseases in which orexin receptors are involved.
Heterobicyclic compounds of Formula (I):
or a pharmaceutically-acceptable salt, tautomer, or stereoisomer thereof, as defined in the specification, and compositions containing them, and processes for preparing such compounds. Provided herein also are methods of treating disorders or diseases treatable by inhibition of PDE10, such as obesity, non-insulin dependent diabetes, schizophrenia, bipolar disorder, obsessive-compulsive disorder, Huntington's Disease, and the like.
Formula (I)的杂环化合物:
或其药用可接受的盐、互变异构体或立体异构体,如规范中所定义,并含有它们的组合物,以及制备这种化合物的方法。本文还提供了通过抑制PDE10来治疗由此可治疗的疾病或疾病的方法,如肥胖症、非胰岛素依赖型糖尿病、精神分裂症、躁郁症、强迫症、亨廷顿病等。
[EN] NAPHTHALENE CARBOXAMIDE M1 RECEPTOR POSITIVE ALLOSTERIC MODULATORS<br/>[FR] COMPOSÉS DE NAPHTHALÈNE CARBOXAMIDE, MODULATEURS ALLOSTÉRIQUES POSITIFS DU RÉCEPTEUR M1
申请人:MERCK SHARP & DOHME
公开号:WO2011149801A1
公开(公告)日:2011-12-01
The present invention is directed to naphthalene carboxamide compounds of formula (I) which are M1 receptor positive allosteric modulators and that are useful in the treatment of diseases in which the M1 receptor is involved, such as Alzheimers disease, schizophrenia, pain or sleep disorders. The invention is also directed to pharmaceutical compositions comprising the compounds and to the use of the compounds and compositions in the treatment of diseases mediated by the M1 receptor.
[EN] COMT INHIBITING METHODS AND COMPOSITIONS<br/>[FR] PROCÉDÉS D'INHIBITION DE LA COMT ET COMPOSITIONS ASSOCIÉES
申请人:LIEBER INST FOR BRAIN DEV
公开号:WO2016123576A1
公开(公告)日:2016-08-04
The present inventions include a method of inhibiting COMT enzyme in a subject as well as compounds of formula I, or a pharmaceutically acceptable salt thereof, that are useful in the treatment of various disorders mediated by COMT, including Parkinson's disease and/or schizophrenia.