
氨基二氟膦 | 25757-74-8
中文名称
氨基二氟膦
中文别名
——
英文名称
aminodifluorophosphine
英文别名
(difluorophosphino)amine;Phosphoramidous difluoride

CAS
25757-74-8
化学式
F2H2NP
mdl
——
分子量
84.9931
InChiKey
MAZAVXNOOKBYCU-UHFFFAOYSA-N
BEILSTEIN
——
EINECS
——
-
物化性质
-
计算性质
-
ADMET
-
安全信息
-
SDS
-
制备方法与用途
-
上下游信息
-
文献信息
-
表征谱图
-
同类化合物
-
相关功能分类
-
相关结构分类
计算性质
-
辛醇/水分配系数(LogP):0.3
-
重原子数:4
-
可旋转键数:0
-
环数:0.0
-
sp3杂化的碳原子比例:0.0
-
拓扑面积:26
-
氢给体数:1
-
氢受体数:3
SDS
上下游信息
反应信息
-
作为反应物:描述:参考文献:名称:Infrared and Raman spectra, conformational stability, structural parameters, ab initio calculations and vibrational assignments of aminodifluorophosphine摘要:Infrared spectra (4000-50 cm(-1)) have been recorded for three isotopomers of aminodifluorophosphine, H2NPF2, (H2NPF2)-N-15 and D2NPF2, of the gases. The Raman spectra of all three molecules were recorded of the liquids. To support the vibrational assignment MP2(full) ab initio calculations with the 6-31G(d) basis set were carried out to predict the fundamental vibrational frequencies, infrared intensities, Raman activities, depolarization values and infrared band contours. Additional ab initio calculations employing a variety of basis sets with and without diffuse functions have been used to predict the conformational stability, structural parameters and centrifugal distortion constants. These predictions are compared to previously reported experimental values when available. The adjusted r(0) structural parameters have been obtained by systematically fitting the MP2(full)/6-311+G(d) predicted values with the previously reported rotational constants for five isotopomers obtained from the microwave study. The difference in the two NH distances is 0.003 angstrom which is much smaller than the value of 0.021 angstrom previously reported. The bonding around the nitrogen atom is effectively planar which is consistent with the previously reported structural information from the microwave study but differs from the reported slightly pyramidal bonding obtained in the electron diffraction investigation. The adjusted r(0) heavy atom distances and angles are: r(PF) = 1.587(3); r(NP) = 1.649(3) angstrom; angle FPF = 94.7(5); angle NPF = 100.6(5)degrees. Vibrational assignments are given for H2NPF2, (H2NPF2)-N-15 and D2NPF2 which are supported by ab initio MP2/6-31G(d) calculations. These results are compared to the corresponding quantities of some similar molecules. (c) 2008 Published by Elsevier B.V.DOI:10.1016/j.molstruc.2007.04.045
-
作为产物:描述:参考文献:名称:Infrared and Raman spectra, conformational stability, structural parameters, ab initio calculations and vibrational assignments of aminodifluorophosphine摘要:Infrared spectra (4000-50 cm(-1)) have been recorded for three isotopomers of aminodifluorophosphine, H2NPF2, (H2NPF2)-N-15 and D2NPF2, of the gases. The Raman spectra of all three molecules were recorded of the liquids. To support the vibrational assignment MP2(full) ab initio calculations with the 6-31G(d) basis set were carried out to predict the fundamental vibrational frequencies, infrared intensities, Raman activities, depolarization values and infrared band contours. Additional ab initio calculations employing a variety of basis sets with and without diffuse functions have been used to predict the conformational stability, structural parameters and centrifugal distortion constants. These predictions are compared to previously reported experimental values when available. The adjusted r(0) structural parameters have been obtained by systematically fitting the MP2(full)/6-311+G(d) predicted values with the previously reported rotational constants for five isotopomers obtained from the microwave study. The difference in the two NH distances is 0.003 angstrom which is much smaller than the value of 0.021 angstrom previously reported. The bonding around the nitrogen atom is effectively planar which is consistent with the previously reported structural information from the microwave study but differs from the reported slightly pyramidal bonding obtained in the electron diffraction investigation. The adjusted r(0) heavy atom distances and angles are: r(PF) = 1.587(3); r(NP) = 1.649(3) angstrom; angle FPF = 94.7(5); angle NPF = 100.6(5)degrees. Vibrational assignments are given for H2NPF2, (H2NPF2)-N-15 and D2NPF2 which are supported by ab initio MP2/6-31G(d) calculations. These results are compared to the corresponding quantities of some similar molecules. (c) 2008 Published by Elsevier B.V.DOI:10.1016/j.molstruc.2007.04.045
-
作为试剂:参考文献:名称:Storzer, Werner; Roeschenthaler, Gerd-Volker; Schmutzler, Reinhard, Chemische Berichte, 1981, vol. 114, # 11, p. 3609 - 3624摘要:DOI:
文献信息
-
Preparation and chemical and spectroscopic properties of (disilylamino)difluorophosphine and bis(difluorophosphino)silylamine作者:E. A. V. Ebsworth、David W. H. Rankin、John G. WrightDOI:10.1039/dt9790001065日期:——N(PF2)2(SiH3) have been prepared by the reactions of SiBrH3 and NMe3 with PF2[NH(SiH3)] and NH(PF2)2 respectively. Vibrational, n.m.r., mass, and photoelectron spectra have been recorded, and chemical properties have been studied. With diborane, PF2[N(SiH3)2] gives the adduct PF2[N(SiH3)2]·BH3 and N(PF2)2(SiH3) gives both mono- and bis-(borane) adducts; n.m.r. spectre of the three borane adducts have
-
Preparation and properties of diaminodifluorophosphorane作者:David E. J. Arnold、David W. H. RankinDOI:10.1039/dt9760001130日期:——Diaminodifluorophosphorane, PF2H(NH2)2, has been prepared by the reaction of chlorodifluorophosphine and ammonia, and has been characterised spectroscopically. Its vibrational spectra can be interpreted in terms of C2ν symmetry, with a trigonal-bipyramidal structure with axial fluorine atoms. N.m.r. spectra are consistent with this structure, and show that the amino-groups rotate rapidly at room temperature
-
Preparation and chemical and spectroscopic properties of bis(difluorophosphino) selenide作者:David E. J. Arnold、Ernest R. Cromie、David W. H. RankinDOI:10.1039/dt9770001999日期:——Bis(difluorophosphino) selenide has been prepared by the exchange reactions of PBrF2 and PCLF2 with Se(SiH3)2 and Se(SnBu3)2. It has been characterised by vrbrational, mass, n.m.r., and photoelectron spectroscopy. Spectroscopic results have also been obtained for bis(difluorophosphino) sulphide, prepared by analogous methods. Both compounds react with Brønsted acids to give PF2HSe or PF2HS, and a difluorophos-phine
-
Preparation and spectroscopic properties of amines containing germyl and difluorophosphino-groups作者:E. A. V. Ebsworth、David W. H. Rankin、John G. WrightDOI:10.1039/dt9770002348日期:——been prepared from the appropriate primary and secondary difluorophosphinoamines by reaction with germyl halide and trimethylamine. The compounds are all much more stable than trigermylamine, but decompose at room temperature by elimination of GeH2. Vibrational, photoelectron, n.m.r., and mass spectroscopic data have been recorded, and are interpreted in terms of the probable conformations adopted by
-
Gas-phase molecular structure of bis(difluorophosphino)amine, determined by electron diffraction作者:Christopher M. Huntley、Graham S. Laurenson、David W. H. RankinDOI:10.1039/dt9800000954日期:——The molecular geometry of bis(difluorophosphino)amine, NH(PF2)2, in the gas phase has been studied by electron diffraction. Principal parameters are: ra(P–F) 158.4(3), ra(P–N) 168.4(8) pm; FPF 95.6(10), FPN 98.3(7), and PN P 122.1(7)°. Two conformers are present in the vapour at room temperature. The predominant form (72%) has almost C2v symmetry, but the PF2 groups are twisted 5°away from most symmetrical
表征谱图
-
氢谱1HNMR
-
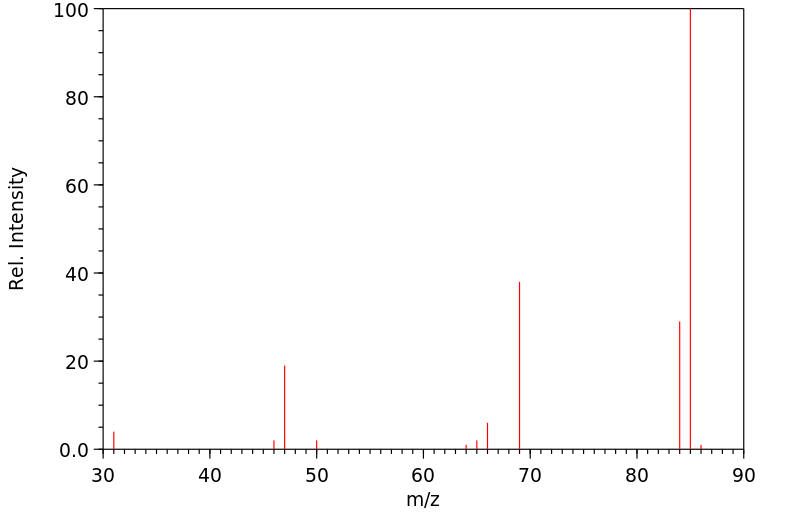
质谱MS
-
碳谱13CNMR
-
红外IR
-
拉曼Raman
-
峰位数据
-
峰位匹配
-
表征信息
同类化合物
磷酰氨基硫杂二氯化(8CI,9CI)
磷酰氨基硫杂二氟化(8CI,9CI)
磷羧基硫杂二溴化(8CI,9CI)
磷羧基亚胺二氯化,N-(氯硫膦基)-
碘化铵
碘二硫醚
硫氯五氟化物
硫氟氧化物
硫单氟化物
硫代磷酰溴
硫二氟化物
环-八(二氟磷酰腈)
溴化铵
溴化硒(IV)
溴化硒
溴化硒
溴(二氯)膦
溴(二氟)膦
氯膦酸
氯胺
氯硒次氯酸盐
氯化铵
氯化硫磷
氯二氟胺
氯-二氟-巯基膦烷
氨基溴
氨基二氟膦
氟氧烷基
氟化铵
氟化磷(III)
氟化氢二聚体阴离子
氟(1+)
次碘(I)酸
次氟酸
次二亚磷二溴化二氯化(Br2ppcl2)(9CI)
四碘化二磷
四氯化硫
四氯化硒
四氯二磷烷
四氟化硫
四氟代肼
双(五氟硫)过氧化物
单氟胺
十氯-1-四磷酸化-1,3,5-三六乙酸氯磷酸酯
六氯环三磷腈
六氟磷酸铵
六氟磷酸盐
六氟磷酸
六氟环三磷腈
六氟化硫
相关结构分类

联系我们



关注我们

公众号

