
4-Hydroxy-2-methyl-3a,4,9,9a-tetrahydro-4,9<1',2'>benzeno-1H-benzisoindole-1,3(2H)-dione | 118494-66-9
分子结构分类
中文名称
——
中文别名
——
英文名称
4-Hydroxy-2-methyl-3a,4,9,9a-tetrahydro-4,9<1',2'>benzeno-1H-benzisoindole-1,3(2H)-dione
英文别名
4-Hydroxy-2-methyl-3a,4,9,9a-tetrahydro-4,9[1',2']benzeno-1H-benz[f]isoindole-1,3(2H)-dione;1-Hydroxy-17-methyl-17-azapentacyclo[6.6.5.02,7.09,14.015,19]nonadeca-2,4,6,9,11,13-hexaene-16,18-dione

CAS
118494-66-9
化学式
C19H15NO3
mdl
——
分子量
305.333
InChiKey
JYJCOMSKUPTVBL-UHFFFAOYSA-N
BEILSTEIN
——
EINECS
——
-
物化性质
-
计算性质
-
ADMET
-
安全信息
-
SDS
-
制备方法与用途
-
上下游信息
-
文献信息
-
表征谱图
-
同类化合物
-
相关功能分类
-
相关结构分类
计算性质
-
辛醇/水分配系数(LogP):1.1
-
重原子数:23
-
可旋转键数:0
-
环数:6.0
-
sp3杂化的碳原子比例:0.26
-
拓扑面积:57.6
-
氢给体数:1
-
氢受体数:3
上下游信息
反应信息
-
作为反应物:参考文献:名称:手性碱催化的不对称Diels-Alder反应摘要:已经发现在催化量的各种手性β-氨基醇的存在下,蒽酮与N-甲基马来酰亚胺反应。以优异的产率获得了光学活性的环加合物3a。已经研究了反应的几个特征。DOI:10.1016/s0040-4039(00)70709-x
-
作为产物:描述:蒽酮 在 palladium on activated charcoal 氢气 作用下, 以 吡啶 为溶剂, 生成 4-Hydroxy-2-methyl-3a,4,9,9a-tetrahydro-4,9<1',2'>benzeno-1H-benz
isoindole-1,3(2H)-dione 参考文献:名称:Anthracenediols as reactive dienes in base-catalyzed cycloadditions: reduction-cycloaddition reactions of anthraquinones摘要:Anthraquinone is readily reduced to the hydroquinone (9,10-anthracenediol), which under basic conditions serves as a reactive diene for cyloaddition purposes. Catalytic hydrogenation in pyridine solvent provides convenient access to this species, and efficient reactions occur with dienophiles in situ, provided that they are sufficiently reactive. Thus N-methylmaleimide (NMM) gives the bicyclic bridgehead diol in near quantitative yield when the H2/Pd reduction of anthraquinone is carried out in pyridine containing 1 equiv of NMM. Fumaronitrile and maleonitrile similarly give high yields in stereospecific reactions, with the dienophile geometry retained in the cycloadduct. Less reactive dienophiles suffer competitive reduction. Dimethyl fumarate in situ gives cycloadduct (stereospecifically) in only 35-60% yield, with the remainder of the dienophile reduced to dimethyl succinate. Stepwise reduction followed by addition of dienophile leads to a higher yield in this and related reactions. The benzologues 5,12-naphthacenedione and 6,13-pentacenedione undergo analogous reactions with NMM, leading to novel bridgehead diols. The monimine of anthraquinone exhibits NMR features attributed to syn/anti isomerism. Under neutral or mildly basic conditions, the aromatic protons on the ring proximal to the NH are clearly distinguished (500 MHz) from those on the distal ring. The addition of acid causes rapid syn/anti NH exchange leading to time averaged symmetry. This imine behaves similarly to anthraquinone in the reduction/cycloaddition sequence. For example, with NMM in situ an essentially quantitative yield of the novel bridgehead amino alcohol adduct is obtained. Related benzologue reactions and attempts to extend the sequence to the oxime and methylene analogues of anthraquinone are described. Base-catalyzed ring opening of the cycloadduct of NMM/anthracenediol leads to a novel retro-bis-aldol reaction, resulting in the formation of anthraquinone and N-methylsuccinimide.DOI:10.1021/jo00004a009
文献信息
-
Asymmetric base-catalyzed cycloaddition between anthrone and some dienophiles作者:Olivier Riant、Henri B. Kagan、Louis RicardDOI:10.1016/s0040-4020(01)89385-6日期:1994.4presence of catalytic amounts of various chiral β-aminoalcohols. The cycloadduct 3a has been obtained in excellent yield with enantiomeric excess of up to 61%. Its absolute configuration has been assigned by X-ray crystallography. Several features of the reaction have been studied: variation of dienophile; competition between cycloaddition and formation of the optically active Michael adduct 4; solvent
-
Base-catalyzed reactions of anthrones with dienophiles作者:Michael Koerner、Bruce RickbornDOI:10.1021/jo00296a024日期:1990.4
-
Anthrones as reactive dienes in Diels-Alder reactions作者:Michael Koerner、Bruce RickbornDOI:10.1021/jo00262a003日期:1989.1
-
RIANT, OLIVIER;KAGAN, HENRI B., TETRAHEDRON LETT., 30,(1989) N2, C. 7403-7406作者:RIANT, OLIVIER、KAGAN, HENRI B.DOI:——日期:——
-
KOERNER, MICHAEL;RICKBORN, BRUCE, J. ORG. CHEM., 55,(1990) N, C. 2662-2672作者:KOERNER, MICHAEL、RICKBORN, BRUCEDOI:——日期:——
表征谱图
-
氢谱1HNMR
-
质谱MS
-
碳谱13CNMR
-
红外IR
-
拉曼Raman
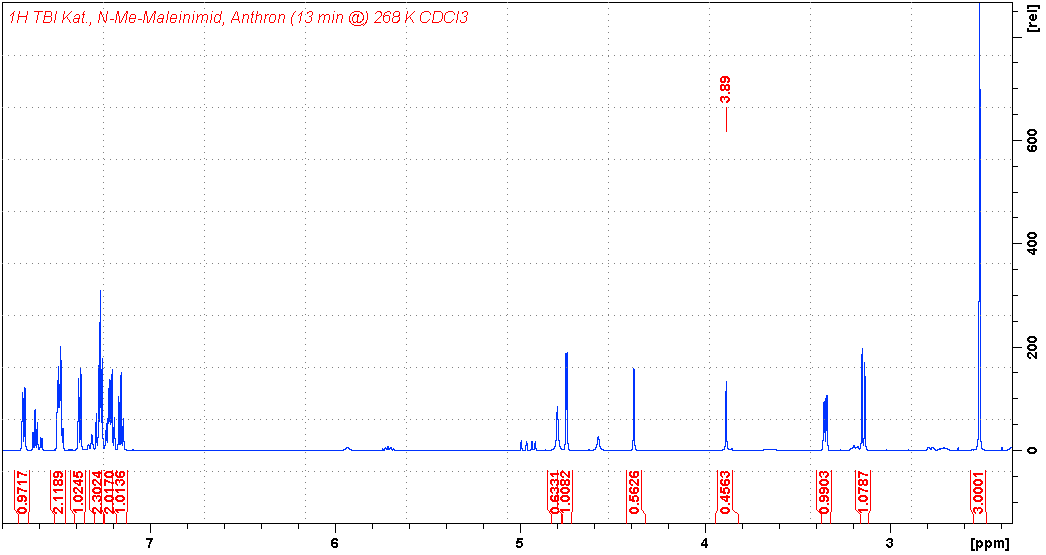
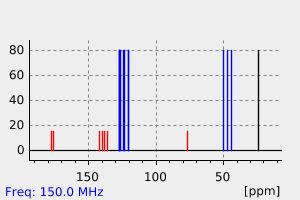
-
峰位数据
-
峰位匹配
-
表征信息
同类化合物
鬼臼酸哌啶基腙氮氧自由基
鬼臼酸
鬼臼毒醇
萘并[2,3-d]-1,3-二噁唑-5(6H)-酮,8-(1,3-苯并二噁唑-5-基)-7,8-二氢-6,7-二甲基-,(6R,7S,8R)-rel-(-)-
萘并[1,2-d]-1,3-二噁唑,9-(1,3-苯并二噁唑-5-基)-6,7-二氢-7,8-二甲基-,(7S)-
苦鬼臼毒醇
米托肼
甘尔布林
珠子草次素
环藤
消泡剂
愈创木素
异落叶松脂素
异紫杉脂素9,9'-缩丙酮
异紫杉脂素
大侧柏酸
四环[6.6.2.02,7.09,14]十六烷-2(7),3,5,9(14),10,12-己烯-15,15,16,16-四甲腈
四氢萘-1,2,3,4-四羧酸四乙酯
叶下珠新素
五脂素A1
9-氰基蒽光二聚体
7,8,9,9-四去氢异落叶松树脂醇
7,14-二氢-7,14-乙桥二苯并[a,h]蒽-15,16-二羧酸二钠盐
7,14-二氢-7,14-乙桥二苯并[a,h]蒽-15,16-二甲酸
6,8-二溴-4-氧代-4H-1-苯并吡喃-3-甲醛
5a-苯基-5a,14c-二氢苯并[a]茚并[2,1-c]芴-5,10-二酮
2,6-亚甲基吖丙因并[2,3-f]异吲哚(9CI)
2,3-萘二甲酰胺,1,2-二氢-7-羟基-1-(4-羟基-3-甲氧苯基)-N2,N3-二[2-(4-羟基苯基)乙基]-6-甲氧基-,(1R,2S)-rel-
1-苯基-1,2,3,4-四氢-萘-2,3-二羧酸
1-(3,4-二羟基苯基)-6,7-二羟基-1,2-二氢萘-2,3-二甲酸
1-(3,4-二甲氧基苯基)-1,2,3,4-四氢-6,7-二甲氧基-2,3-萘二甲醇
1-(3,4-二甲氧基-苯基)-6,7-二甲氧基-1,2,3,4-四氢-萘-2,3-二羧酸
1-(3,4-二甲氧基-苯基)-6,7-二甲氧基-1,2,3,4-四氢-萘-2,3-二羧酸
(7S,8S,9R)-9-(3,4-二甲氧基苯基)-6,7,8,9-四氢-4-甲氧基-7,8-双(甲氧基甲基)萘并[1,2-D]-1,3-二恶茂
(7S,8R,9R)-9-(1,3-苯并二氧戊环-5-基)-7,8-二甲基-6,7,8,9-四氢苯并[g][1,3]苯并二氧戊环
(1S,2R,3S)-1-(3,4-二甲氧基苯基)-1,2,3,4-四氢-6,7-二甲氧基-2,3-二甲基-萘
(11S,12R)-9,10-乙桥-9,10-二氢蒽-11,12-二甲酸
(-)-南烛木树脂酚
(+)-异落叶松脂素
1-Benzyl-2-oxo-indan-1-carboxylic acid methyl ester
anti-5,6,11,12,14,15,17,18-octahydro-13H,16H-5,12:6,11-di-endo-cyclopropatetracene-15,18-dimethanol bis(methanesulfonate)
7-Hydroxymethyl-5-(3,4,5-trimethoxy-phenyl)-7,8-dihydro-5H-naphtho[2,3-d][1,3]dioxole-6,6-dicarboxylic acid
2-(11-hydroxy-9,10-dihydro-9,10-ethanoanthracen-11-yl)ethyl 4-methylbenzenesulfonate
N-acetyl-N-(13,16-dioxo-9,10-dihydro-9,10-[1,2]benzenoanthracen-1-yl)acetamide
3,3'-((((11S,12S)-9,10-dihydro-9,10-ethanoanthracene-11,12-diyl)bis(azanediyl))bis(methylene))bis(benzene-1,2-diol)
4-(3,4-Dimethoxyphenyl)-5-(3-methoxy-2-hydroxyphenyl)-1-p-tolyl-2,3-pyrrolidindion
2,3,5,6-tetramethoxy-1,4-dihydro-1,4-epoxynaphthalene
9,10-Dimethoxy-9,10-anthraceno-9,10,11,16-benzoanthracen

联系我们



关注我们

公众号
