
1-(4-溴苯基)哌嗪盐酸盐 | 68104-62-1
中文名称
1-(4-溴苯基)哌嗪盐酸盐
中文别名
——
英文名称
1-(4-bromophenyl)piperazine hydrochloride
英文别名
1-(4-Bromophenyl)Piperazine Monohydrochloride;1-(4-bromophenyl)piperazine;hydrochloride

CAS
68104-62-1
化学式
C10H13BrN2*ClH
mdl
MFCD00674134
分子量
277.592
InChiKey
YDVSFRZKQMQPJD-UHFFFAOYSA-N
BEILSTEIN
——
EINECS
——
-
物化性质
-
计算性质
-
ADMET
-
安全信息
-
SDS
-
制备方法与用途
-
上下游信息
-
文献信息
-
表征谱图
-
同类化合物
-
相关功能分类
-
相关结构分类
物化性质
-
熔点:>280°C (dec.)
-
溶解度:DMSO:10mg/mL; PBS(pH 7.2):10 mg/mL
计算性质
-
辛醇/水分配系数(LogP):2.31
-
重原子数:14
-
可旋转键数:1
-
环数:2.0
-
sp3杂化的碳原子比例:0.4
-
拓扑面积:19.8
-
氢给体数:1
-
氢受体数:2
安全信息
-
危险品标志:Xi
-
安全说明:S26,S36/37/39
-
危险类别码:R36/37/38
-
海关编码:2933599090
-
储存条件:存储条件:室温且干燥环境中
制备方法与用途
应用
苯基哌嗪类化合物具有不同程度的5-HT受体阻断活性。当将该部分与其他部分载体结合时,可以使化合物具备中枢神经调节作用或中枢及外周降压活性。
1-(4-溴苯基)哌嗪盐酸盐是苯基哌嗪类抗抑郁症药物的关键中间体,尤其在合成如阿立哌唑等调节神经类药物中扮演重要角色。
制备
以二乙醇胺为起始原料,在无溶剂条件下进行氯代反应生成β, β′-二氯代二乙基胺盐酸盐。不经分离直接滴加取代苯胺和相转移催化剂,经环合反应合成目标化合物1-(4-溴苯基)哌嗪盐酸盐。
反应信息
-
作为反应物:描述:1-(4-溴苯基)哌嗪盐酸盐 在 dimethyl sulfide borane 、 三乙胺 作用下, 以 四氢呋喃 、 二氯甲烷 为溶剂, 反应 22.0h, 生成 1-(4-bromophenyl)-4-(2,2,2-trifluoroethyl)piperazine参考文献:名称:PYRROLO-AROMATIC HETEROCYCLIC COMPOUND, PREPARATION METHOD THEREFOR, AND MEDICAL USE THEREOF摘要:公开号:EP3556761B1
-
作为产物:描述:参考文献:名称:[EN] 6-SUBSTITUTED PYRIDAZINE COMPOUNDS AS SMARCA2 AND/OR SMARCA4 DEGRADERS
[FR] COMPOSÉS DE PYRIDAZINE SUBSTITUÉS EN POSITION 6 SERVANT D'AGENTS DE DÉGRADATION DE SMARCA2 ET/OU SMARCA4摘要:本发明提供了式(I)的6-取代吡啶嗪化合物,作为SMARCA2和/或SMARCA4蛋白的降解剂,在治疗和/或延缓依赖于SMARCA2和/或SMARCA4的疾病或紊乱的进展方面具有治疗作用。这些化合物在制备中也具有用途,以及包含至少一种式(I)化合物或其药用可接受盐、立体异构体、互变异构体或前药的药物组合物。公开号:WO2022029617A1
文献信息
-
[EN] POLYCYCLIC COMPOUNDS AND METHODS FOR THE TARGETED DEGRADATION OF RAPIDLY ACCELERATED FIBROSARCOMA POLYPEPTIDES<br/>[FR] COMPOSÉS POLYCYCLIQUES ET MÉTHODES POUR LA DÉGRADATION CIBLÉE DE POLYPEPTIDES DU FIBROSARCOME RAPIDEMENT ACCÉLÉRÉ申请人:ARVINAS OPERATIONS INC公开号:WO2020051564A1公开(公告)日:2020-03-12The present disclosure relates to bifunctional compounds, ULM— L—PTM, which find utility as modulators of Rapidly Accelerated Fibrosarcoma (RAF, such as c-RAF, A- RAF and/or B-RAF; the target protein). In particular, the present disclosure is directed to bifunctional compounds, which contain on one end a Von Hippel-Lindau, cereblon, Inhibitors of Apotosis Proteins or mouse double-minute homolog 2 ligand which binds to the respective E3 ubiquitin ligase and on the other end a moiety which binds the target protein RAF, such that the target protein is placed in proximity to the ubiquitin ligase to effect degradation (and inhibition) of target protein. The present disclosure exhibits a broad range of pharmacological activities associated with degradation/inhibition of target protein. Diseases or disorders that result from aggregation or accumulation of the target protein, or the constitutive activation of the target protein, are treated or prevented with compounds and compositions of the present disclosure.
-
1-biaryl-1,8-naphthyridin-4-one phosphodiesterase-4 inhibitors
-
A Novel Class of Nonpeptidic Biaryl Inhibitors of Human Cathepsin K作者:Joël Robichaud、Renata Oballa、Peppi Prasit、Jean-Pierre Falgueyret、M. David Percival、Gregg Wesolowski、Sevgi B. Rodan、Donald Kimmel、Colena Johnson、Cliff Bryant、Shankar Venkatraman、Eduardo Setti、Rohan Mendonca、James T. PalmerDOI:10.1021/jm0301078日期:2003.8.1identification of compound (R)-2, a potent human cathepsin K inhibitor (IC(50) = 3 nM) that is selective versus cathepsins B (IC(50) = 3950 nM), L (IC(50) = 3725 nM), and S (IC(50) = 2010 nM). In an in vitro assay involving rabbit osteoclasts and bovine bone, compound (R)-2 inhibited bone resorption with an IC(50) of 95 nM. It was shown that, unlike some peptidic nitrile inhibitors of cysteine proteases, the一种新的非肽联芳基化合物系列被鉴定为组织蛋白酶K的有效抑制剂和可逆抑制剂。已知氨基乙腈二肽1的P2-P3酰胺键被苯环取代,从而形成了与组织蛋白酶相比仍保持效力的联芳基系列K和对其他组织蛋白酶显示出改善的选择性。该系列中的结构修饰导致鉴定了化合物(R)-2,这是一种有效的人组织蛋白酶K抑制剂(IC(50)= 3 nM),相对于组织蛋白酶B(IC(50)= 3950 nM),L( IC(50)= 3725 nM),S(IC(50)= 2010 nM)。在涉及兔破骨细胞和牛骨的体外测定中,化合物(R)-2以95 nM的IC(50)抑制骨吸收。结果表明,与某些半胱氨酸蛋白酶的肽腈抑制剂不同,组织蛋白酶K不会将(R)-2的腈部分转化为相应的酰胺3。这表明这类非肽腈抑制剂不太可能被半胱氨酸蛋白酶水解。此外,显示化合物(R)-2对组织蛋白酶K的抑制是完全可逆的,并且不是时间依赖性的。为了证明化合物(R)-2在体内的功效,将其以20
-
MMP-13-SELECTIVE INHIBITOR申请人:SHIONOGI & CO., LTD.公开号:EP1997804A1公开(公告)日:2008-12-03Since it is thought that if the activity of MMP-13 can be inhibited, this will largely contribute to improvement or prevention of progession of pathological states, particularly, osteoarthritis (OA), resulting from or associated with the MMP-13 activity, the development of MMP-13 inhibitors is anticipated. There are provided a compound represented by the general formula (I): wherein R1 is optionally substituted aryl etc. Z is C1-C5 alkylene which may be substituted and may be interrupted with a substituent selected from Substituent group a etc.; A is the formula: (R6 and R7 are each independently halogen, lower alkyl etc.; m and n are each independently 0, 1, or 2); R2 is a hydrogen atom, optionally substituted lower alkyl etc.; R3 is a hydrogen atom, optionally substituted lower alkyl etc.; R4 is a hydrogen atom; or R3 and R4 may be taken together with an adjacent carbon atom to from a ring; R5 is hydroxy, lower alkyloxy etc.), or an optically active isomer, a pharmaceutically acceptable salt or a solvate thereof, and a pharmaceutical composition containing it as an active ingredient, which has the MMP-13 inhibiting activity.由于人们认为如果MMP-13的活性可以被抑制,这将在改善或预防病理状态的进展方面起到很大作用,特别是由于或与MMP-13活性相关的骨关节炎(OA),因此预期会开发MMP-13抑制剂。 提供了一个由通式(I)表示的化合物: 其中R1是可选择取代的芳基等。Z是可取代的C1-C5烷基,可以被取代并且可以被来自取代基团a等的取代基中断;A是式: (R6和R7各自独立地是卤素,较低烷基等;m和n各自独立地是0、1或2);R2是氢原子,可选择取代的较低烷基等;R3是氢原子,可选择取代的较低烷基等;R4是氢原子;或R3和R4可以与相邻的碳原子一起形成环;R5是羟基,较低烷氧基等),或其光学活性异构体、药学上可接受的盐或其溶剂化合物,以及含有其作为活性成分的具有MMP-13抑制活性的药物组合物。
-
Exploration of substituted arylpiperazine–tetrazoles as promising dual norepinephrine and dopamine reuptake inhibitors作者:Suresh Paudel、Srijan Acharya、Goo Yoon、Kyeong-Man Kim、Seung Hoon CheonDOI:10.1016/j.bmc.2016.09.005日期:2016.11In the search for potent dual norepinephrine and dopamine reuptake inhibitors, several substituted arylpiperazine–tetrazoles were designed, synthesized and evaluated for their neurotransmitter reuptake inhibitory activities. Various derivatives exhibited selective and strong neurotransmitter reuptake inhibitory activity. In particular, compounds with a three-carbon linker displayed selective and stronger
表征谱图
-
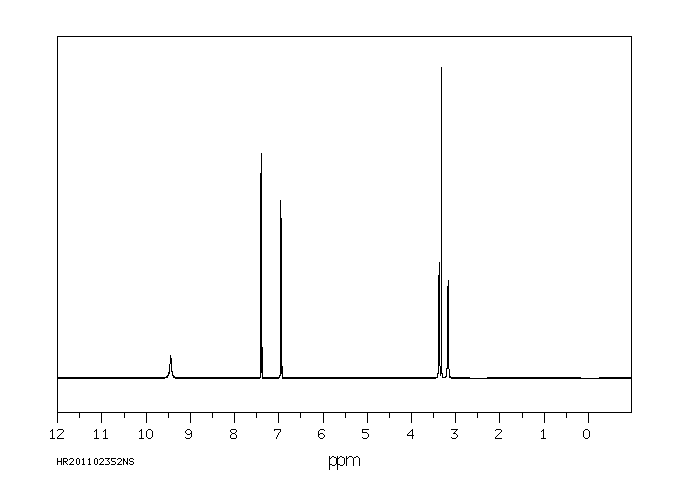
氢谱1HNMR
-
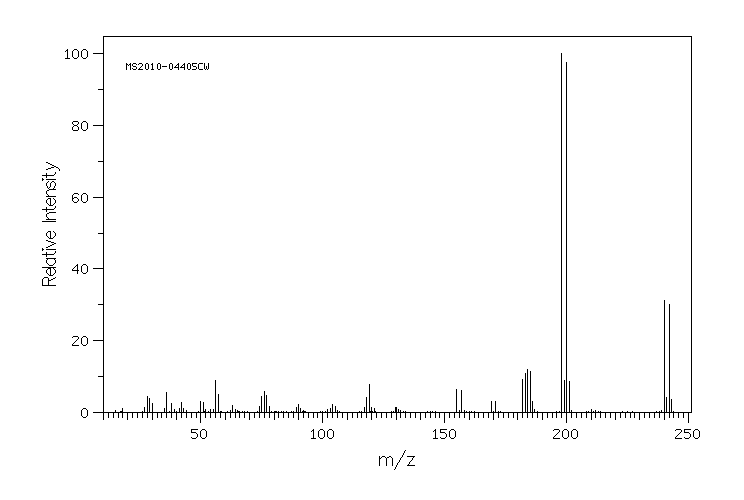
质谱MS
-
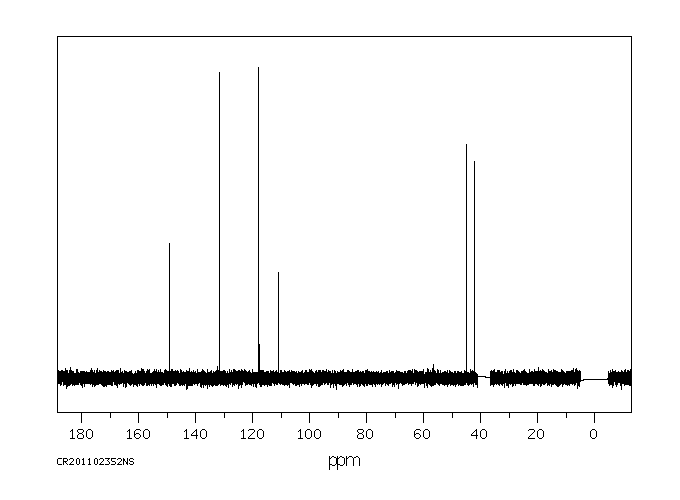
碳谱13CNMR
-
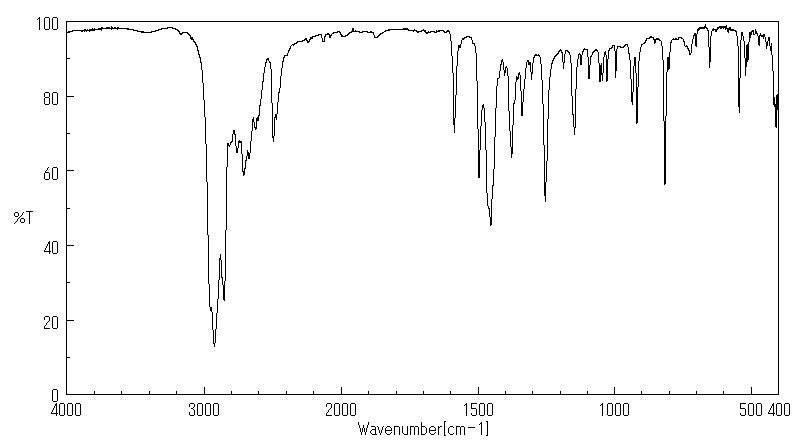
红外IR
-
拉曼Raman
-
峰位数据
-
峰位匹配
-
表征信息
同类化合物
(2S)-4-[7-(8-氯-1-萘)-5,6,7,8-四氢-2-[[((2S)-1-甲基-2-吡咯烷基]甲氧基]吡啶基[3,4-d]嘧啶-4-基]-1-(2-氟-1-氧代-2-丙烯-1-基)-2-哌Chemicalbook嗪乙腈;2-((S)-4-(7-(8-氯萘-1-基)-2-((((S)-1-
齐拉西酮相关物质C
齐拉西酮杂质E
齐拉西酮开环物,氨基酸杂质
齐拉西酮亚砜
齐拉西酮 盐酸盐 一水合物
齐拉西酮
鲸蜡硬脂醇
鲁拉西酮杂质3
鲁拉西酮杂质23
鲁拉西酮杂质14
鲁拉西酮杂质11
鲁拉西酮
鲁拉西杂质E
鲁拉西杂质1
高分子量聚合三嗪类无卤阻燃剂
驱虫灵D
马福拉嗪
马来酸阿伐曲泊帕
顺式-1-乙酰基-2,6-二甲基-4-亚硝基-哌嗪
雷诺嗪双(N-氧化物)
陶扎色替
阿达色林
阿莫西林二氧代哌嗪
阿立哌唑羟基丁基杂质
阿立哌唑杂质26
阿立哌唑USP相关物质H
阿立哌唑N4-氧化物
阿立哌唑N,N-二氧化物
阿立哌唑EP杂质D
阿立哌唑-d8
阿立哌唑
阿泊替尼
阿替韦啶
阿拉诺丁
阿扎哌醇
阿得巴司
阿尔哌汀
阿伐曲泊帕杂质
间羟基苯基哌嗪
钾 1-甲基-4-三氟硼酸三甲基哌嗪
钠4-(4-乙酰基-1-哌嗪基)苯酚
酮齐拉西酮
酮酮唑油酸酯
酚酞单磷酸酯二-(2-氨基-2-甲基-1,3-丙二醇)盐
选择性氟试剂II
达鲁舍替
达哌唑
赫普索
赤霉素A7甲酯

联系我们



关注我们

公众号

